

Syndromic ciliopathy: a taiwanese single-center study
- PMID: 38671463
- PMCID: PMC11046915
- DOI: 10.1186/s12920-024-01880-0
Syndromic ciliopathy: a taiwanese single-center study
Abstract
Background: Syndromic ciliopathies are a group of congenital disorders characterized by broad clinical and genetic overlap, including obesity, visual problems, skeletal anomalies, mental retardation, and renal diseases. The hallmark of the pathophysiology among these disorders is defective ciliary functions or formation. Many different genes have been implicated in the pathogenesis of these diseases, but some patients still remain unclear about their genotypes.
Methods: The aim of this study was to identify the genetic causes in patients with syndromic ciliopathy. Patients suspected of or meeting clinical diagnostic criteria for any type of syndromic ciliopathy were recruited at a single diagnostic medical center in Southern Taiwan. Whole exome sequencing (WES) was employed to identify their genotypes and elucidate the mutation spectrum in Taiwanese patients with syndromic ciliopathy. Clinical information was collected at the time of patient enrollment.
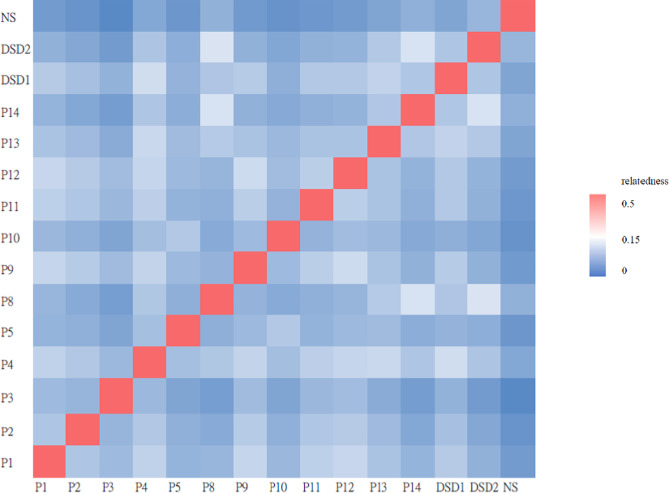
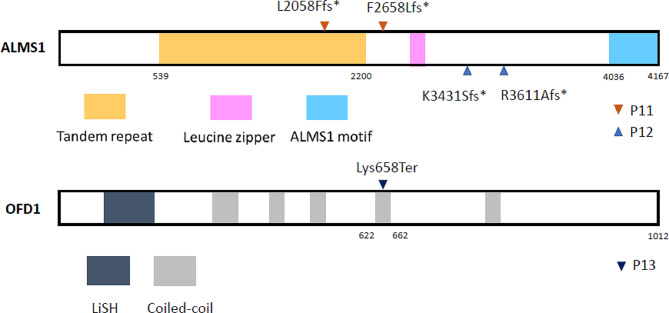
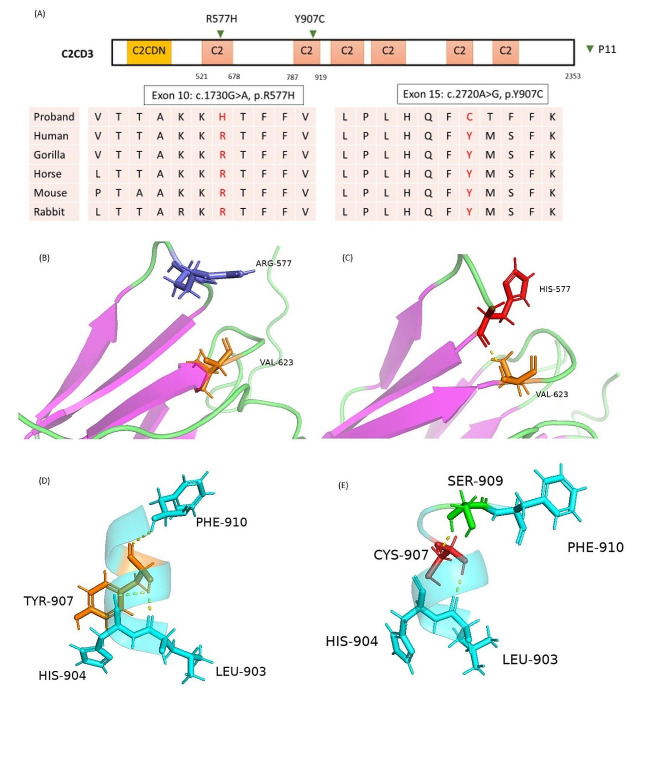
Results: A total of 14 cases were molecularly diagnosed with syndromic ciliopathy. Among these cases, 10 had Bardet-Biedl syndrome (BBS), comprising eight BBS2 patients and two BBS7 patients. Additionally, two cases were diagnosed with Alström syndrome, one with Oral-facial-digital syndrome type 14, and another with Joubert syndrome type 10. A total of 4 novel variants were identified. A recurrent splice site mutation, BBS2: c.534 + 1G > T, was present in all eight BBS2 patients, suggesting a founder effect. One BBS2 patient with homozygous c.534 + 1G > T mutations carried a third ciliopathic allele, TTC21B: c.264_267dupTAGA, a nonsense mutation resulting in a premature stop codon and protein truncation.
Conclusions: Whole exome sequencing (WES) assists in identifying molecular pathogenic variants in ciliopathic patients, as well as the genetic hotspot mutations in specific populations. It should be considered as the first-line genetic testing for heterogeneous disorders characterized by the involvement of multiple genes and diverse clinical manifestations.
Keywords: Alström syndrome; Bardet-biedl syndrome; Ciliopathy; Joubert syndrome; Oral-facial-digital syndrome; Whole exome sequencing.
© 2024. The Author(s).
Conflict of interest statement
Competing interests. The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
References
Publication types
MeSH terms
Substances
Supplementary concepts
LinkOut - more resources
Full Text Sources
Medical
