

A novel COL4A5 splicing mutation causes alport syndrome in a Chinese family
- PMID: 38671472
- PMCID: PMC11046743
- DOI: 10.1186/s12920-024-01878-8
A novel COL4A5 splicing mutation causes alport syndrome in a Chinese family
Abstract
Background: Alport syndrome (AS) is characterised by haematuria, proteinuria, a gradual decline in kidney function, hearing loss, and eye abnormalities. The disease is caused by mutations in COL4An (n = 3, 4, 5) that encodes 3-5 chains of type IV collagen in the glomerular basement membrane. AS has three genetic models: X-linked, autosomal recessive, and autosomal dominant. The most common type of AS is X-linked AS, which is caused by COL4A5.
Methods: We enrolled children with renal insufficiency and a family history of kidney disorders. The proband was identified using whole-exome sequencing. Sanger sequencing was performed to verify the mutation site. Minigene technology was used to analyse the influence of mutant genes on pre-mRNA shearing, and the Iterative Threading ASSEmbly Refinement (I-TASSER) server was used to analyse the protein structure changes.
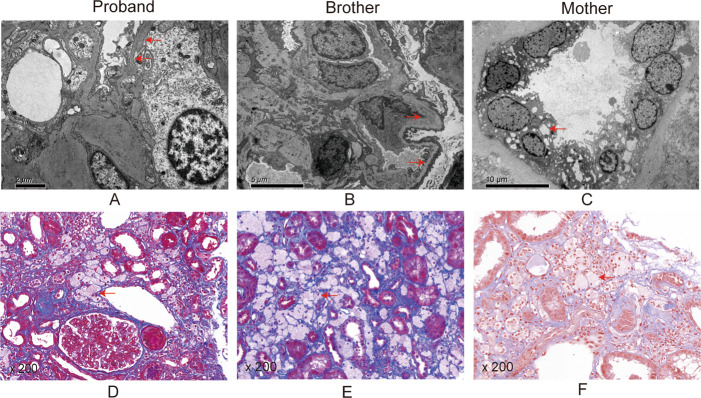
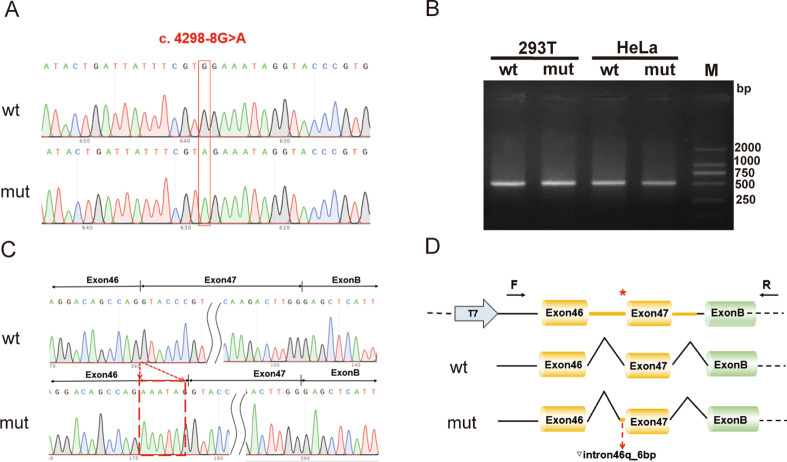
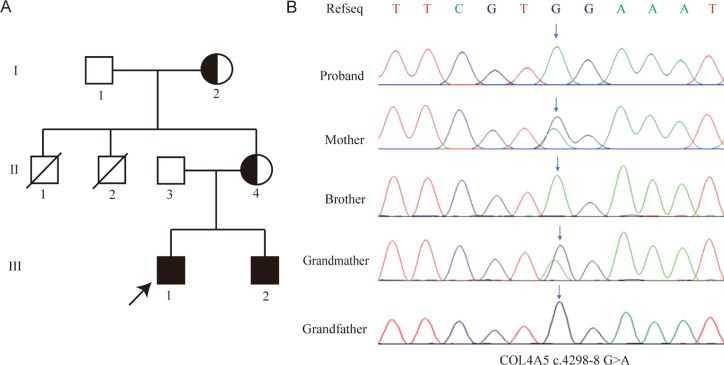
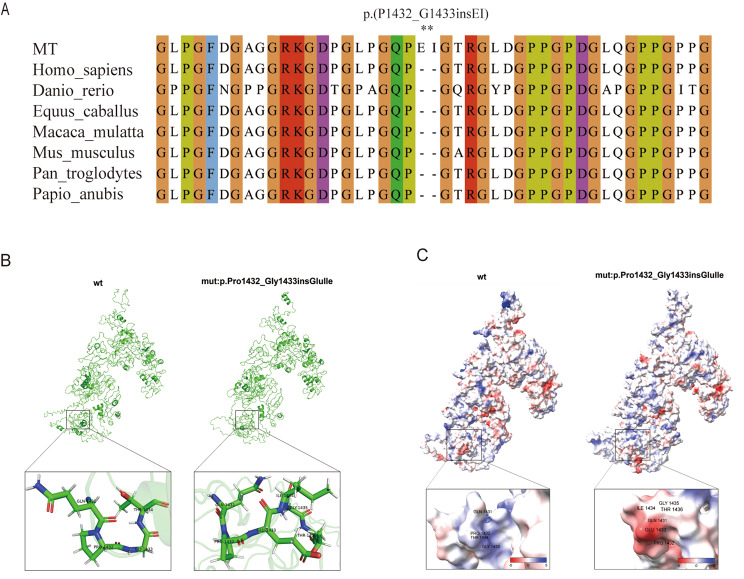
Results: The proband, together with her mother and younger brother, displayed microscopic haematuria and proteinuria, Pathological examination revealed mesangial hyperplasia and sclerosis. A novel mutation (NM_000495.5 c.4298-8G > A) in the intron of the COL4A5 gene in the proband was discovered, which was also present in the proband's mother, brother, and grandmother. In vitro minigene expression experiments verified that the c.4298-8G > A mutation caused abnormal splicing, leading to the retention of six base pairs at the end of intron 46. The I-TASSER software predicted that the mutation affected the hydrogen-bonding structure of COL4A5 and the electrostatic potential on the surface of the protein molecules.
Conclusions: Based on the patient's clinical history and genetic traits, we conclude that the mutation at the splicing site c.4298-8G > A of the COL4A5 gene is highly probable to be the underlying cause within this particular family. This discovery expands the genetic spectrum and deepens our understanding of the molecular mechanisms underlying AS.
Keywords: COL4A5 gene; Aberrant splicing; Alport syndrome; Minigene assay.
© 2024. The Author(s).
Conflict of interest statement
Competing interests. The authors declare no competing interests.
Figures
Similar articles
-
A comprehensive splicing characterization of COL4A5 mutations and prognostic significance in a single cohort with X-linked alport syndrome.Front Genet. 2025 Jun 11;16:1564343. doi: 10.3389/fgene.2025.1564343. eCollection 2025. Front Genet. 2025. PMID: 40567900 Free PMC article.
-
Unveiling Maternal Germline Mosaicism in X-Linked Alport Syndrome by Advanced Genetic Testing.Am J Case Rep. 2025 Aug 6;26:e947287. doi: 10.12659/AJCR.947287. Am J Case Rep. 2025. PMID: 40765127 Free PMC article.
-
De novo variation in ARID1B gene causes Coffin-Siris syndrome 1 in a Chinese family with excessive early-onset high myopia.BMC Med Genomics. 2024 May 24;17(1):142. doi: 10.1186/s12920-024-01904-9. BMC Med Genomics. 2024. PMID: 38790056 Free PMC article.
-
Genotype-Based Molecular Mechanisms in Alport Syndrome.J Am Soc Nephrol. 2025 Jun 1;36(6):1176-1183. doi: 10.1681/ASN.0000000647. Epub 2025 Feb 3. J Am Soc Nephrol. 2025. PMID: 39899372 Review.
-
Potential Renal Damage Biomarkers in Alport Syndrome-A Review of the Literature.Int J Mol Sci. 2022 Jun 30;23(13):7276. doi: 10.3390/ijms23137276. Int J Mol Sci. 2022. PMID: 35806283 Free PMC article.
References
-
- Coppo R, Gianoglio B, Porcellini M, Maringhini S. Frequency of renal diseases and clinical indications for renal biopsy in children (report of the Italian National Registry of Renal biopsies in Children). Group of Renal Immunopathology of the Italian Society of Pediatric Nephrology and Group of Renal Immunopathology of the Italian Society of Nephrology. Nephrology, dialysis, transplantation: official publication of the European Dialysis and Transplant Association -. Eur Ren Association. 1998;13(2):293–7. 10.1093/oxfordjournals.ndt.a027821. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
