

Single-Center Experience of Pediatric Cystic Kidney Disease and Literature Review
- PMID: 38671609
- PMCID: PMC11048964
- DOI: 10.3390/children11040392
Single-Center Experience of Pediatric Cystic Kidney Disease and Literature Review
Abstract
Introduction: Pediatric cystic kidney disease (CyKD) includes conditions characterized by renal cysts. Despite extensive research in this field, there are no reliable genetics or other biomarkers to estimate the phenotypic consequences. Therefore, CyKD in children heavily relies on clinical and diagnostic testing to predict the long-term outcomes.
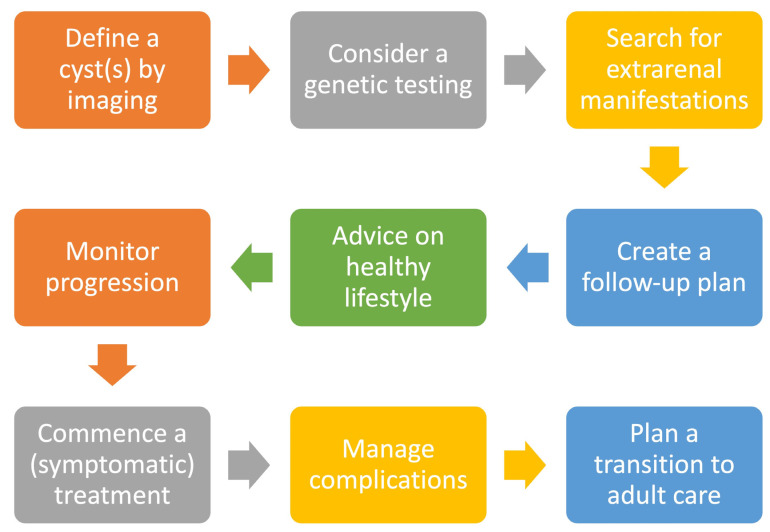
Aim: A retrospective study aimed to provide a concise overview of this condition and analyze real-life data from a single-center pediatric CyKD cohort followed during a 12-year period.
Methods and materials: Medical records were reviewed for extensive clinical, laboratory, and radiological data, treatment approaches, and long-term outcomes.
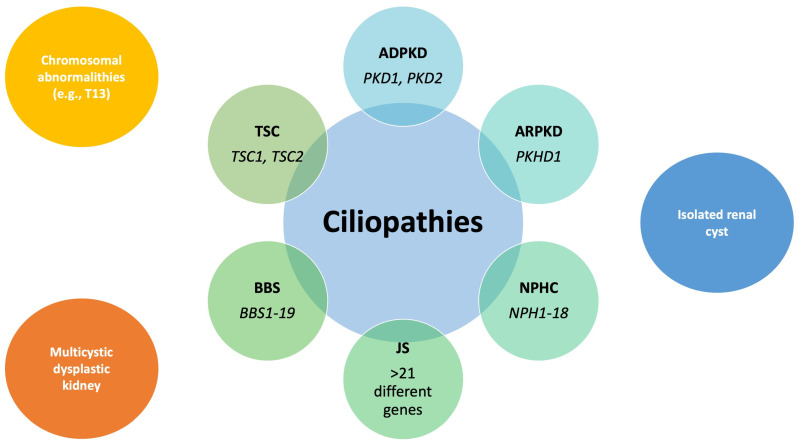
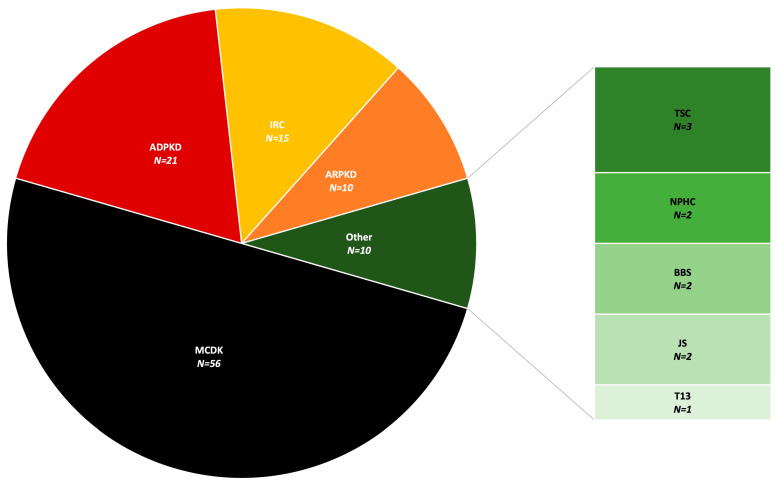
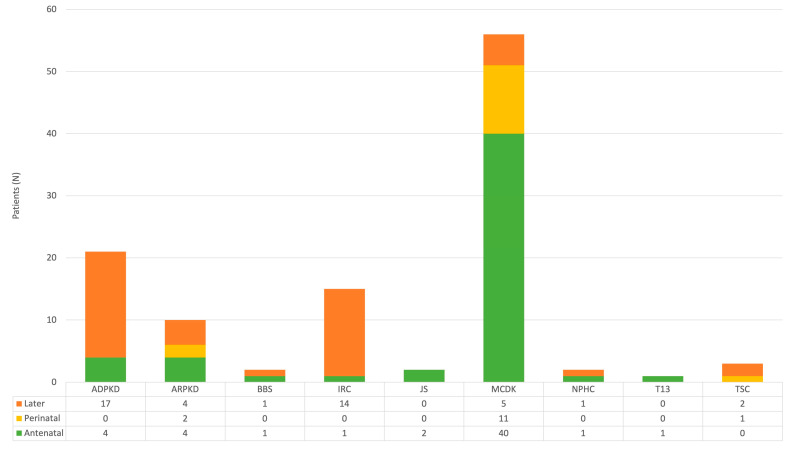
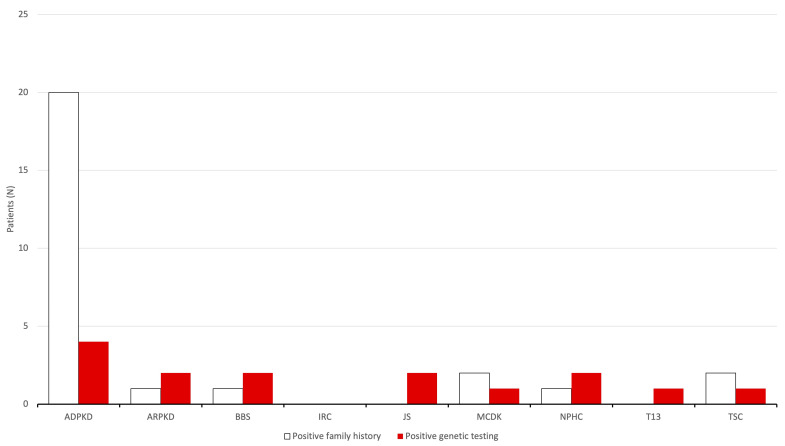
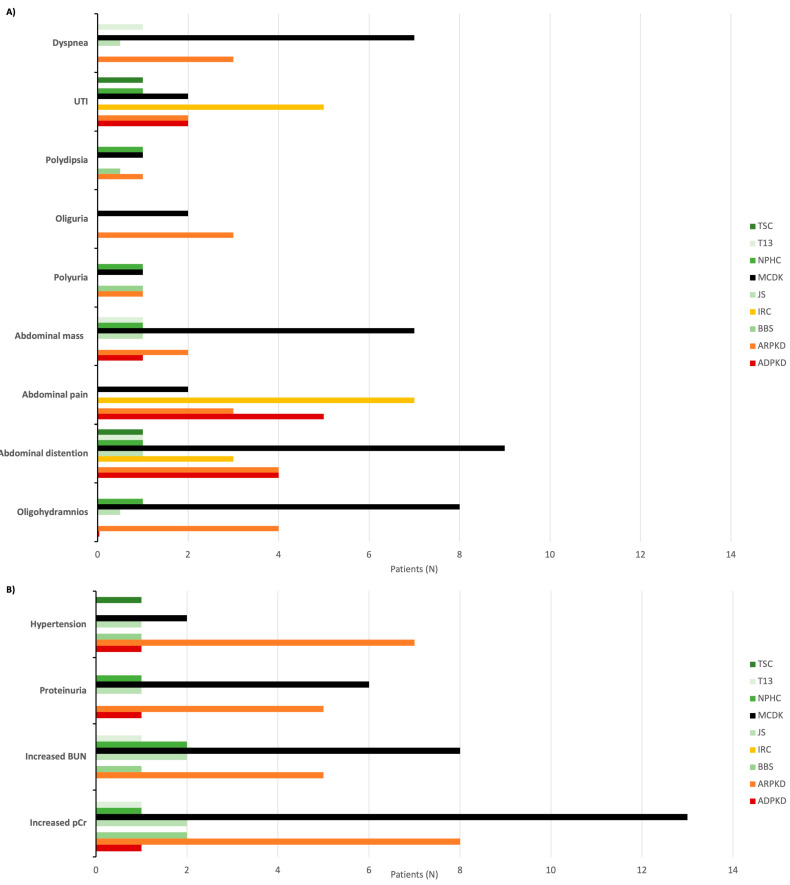
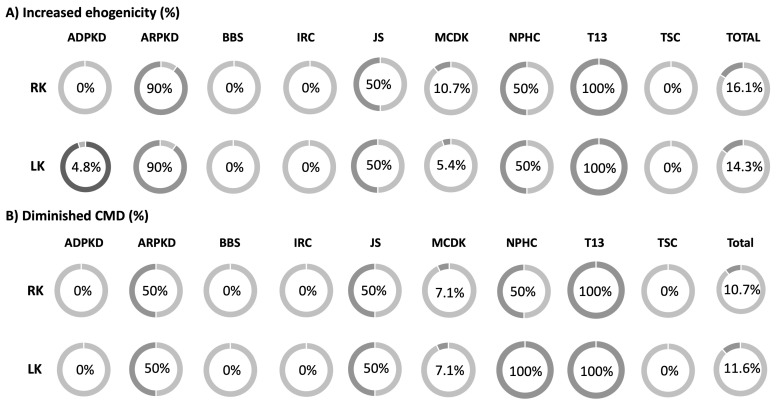
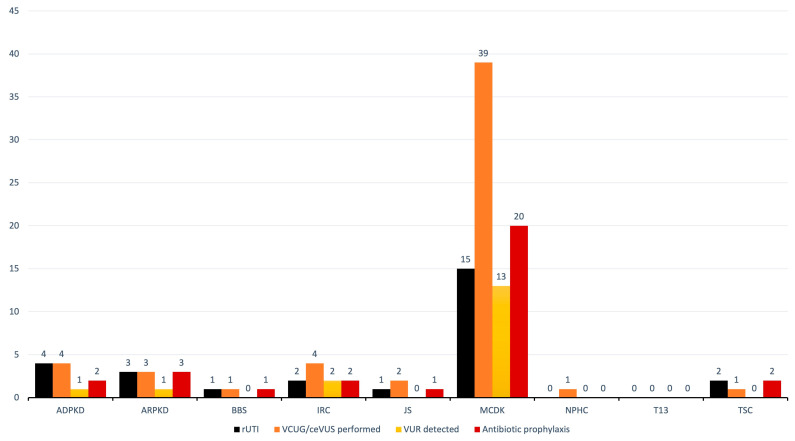
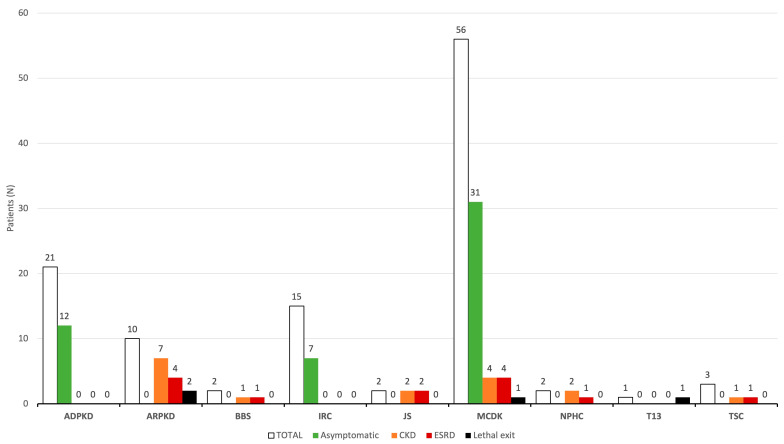
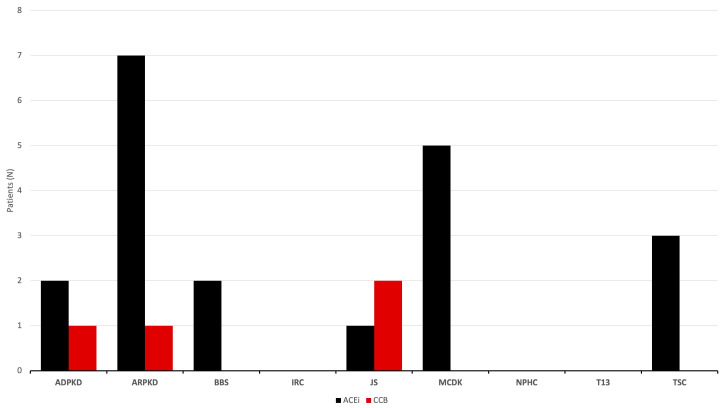
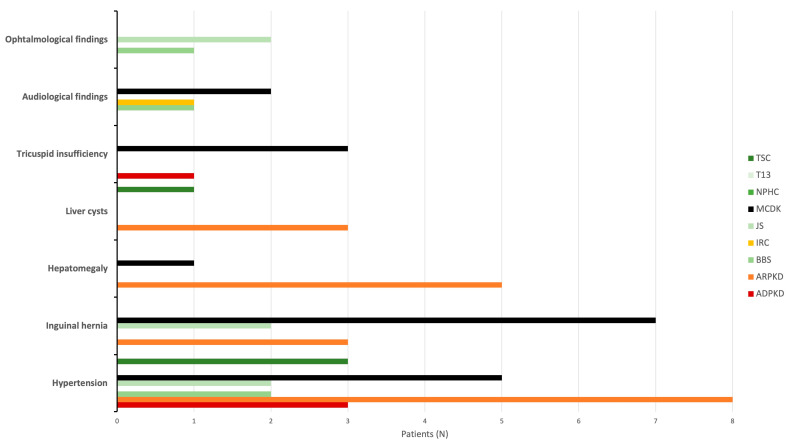
Results: During the study period, 112 patients received a diagnosis of pediatric CyKD. Male patients were more involved than female (1:0.93). Fifty-six patients had a multicystic dysplastic kidney; twenty-one of them had an autosomal dominant disorder; fifteen had an isolated renal cyst; ten had been diagnosed with autosomal recessive polycystic kidney disease; three had the tuberous sclerosis complex; two patients each had Bardet-Biedl, Joubert syndrome, and nephronophthisis; and one had been diagnosed with the trisomy 13 condition. Genetic testing was performed in 17.9% of the patients, revealing disease-causing mutations in three-quarters (75.0%) of the tested patients. The most commonly presenting symptoms were abdominal distension (21.4%), abdominal pain (15.2%), and oligohydramnios (12.5%). Recurrent urinary tract infections (UTI) were documented in one-quarter of the patients, while 20.5% of them developed hypertension during the long-term follow-up. Antibiotic prophylaxis and antihypertensive treatment were the most employed therapeutic modalities. Seventeen patients progressed to chronic kidney disease (CKD), with thirteen of them eventually reaching end-stage renal disease (ESRD). The time from the initial detection of cysts on an ultrasound (US) to the onset of CKD across the entire cohort was 59.0 (7.0-31124.0) months, whereas the duration from the detection of cysts on an US to the onset of ESRD across the whole cohort was 127.0 (33.0-141.0) months. The median follow-up duration in the cohort was 3.0 (1.0-7.0) years. The patients who progressed to ESRD had clinical symptoms at the time of initial clinical presentation.
Conclusion: This study is the first large cohort of patients reported from Croatia. The most common CyKD was the multicystic dysplastic kidney disease. The most common clinical presentation was abdominal distention, abdominal pain, and oliguria. The most common long-term complications were recurrent UTIs, hypertension, CKD, and ESRD.
Keywords: ADPKD; ARPKD; Bardet–Biedl syndrome; Joubert syndrome; cystic kidney disease; multicystic dysplastic kidney; nephronophthisis complex; tuberous sclerosis complex.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
References
LinkOut - more resources
Full Text Sources
