

Cellular and Molecular Mechanisms of Heterotopic Ossification in Fibrodysplasia Ossificans Progressiva
- PMID: 38672135
- PMCID: PMC11048698
- DOI: 10.3390/biomedicines12040779
Cellular and Molecular Mechanisms of Heterotopic Ossification in Fibrodysplasia Ossificans Progressiva
Abstract
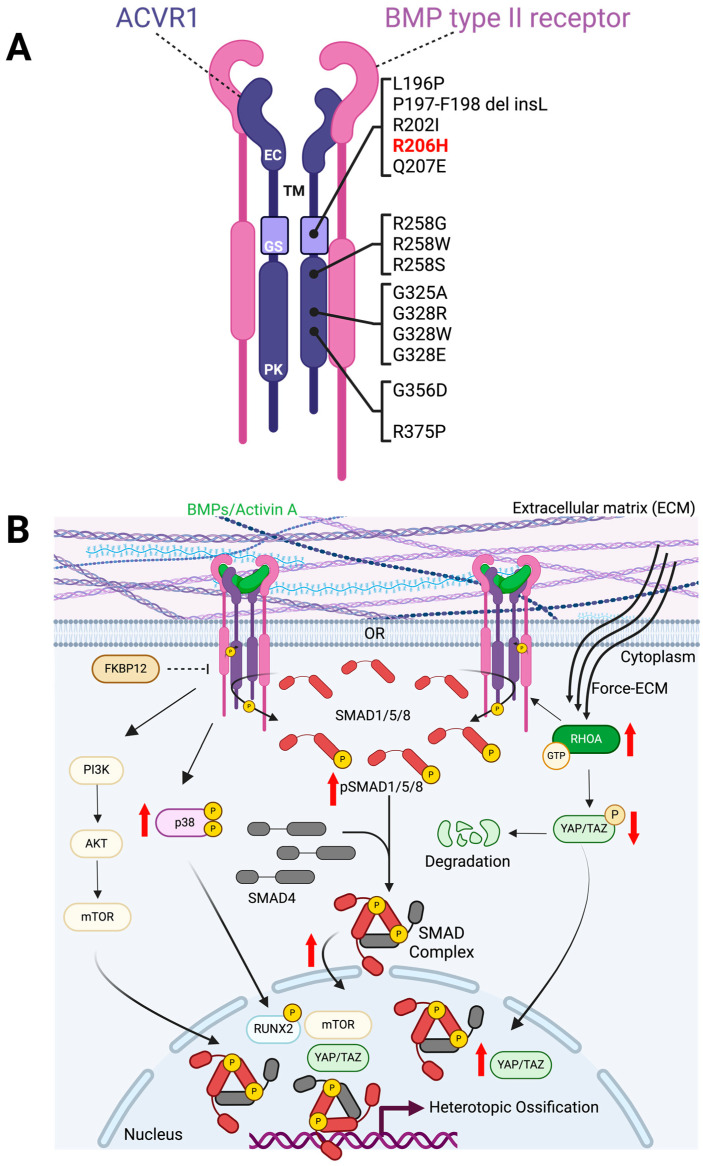
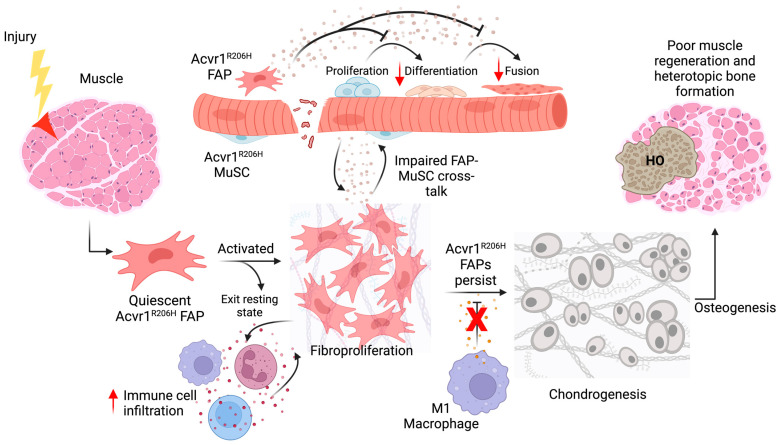
Fibrodysplasia ossificans progressiva (FOP) is a debilitating genetic disorder characterized by recurrent episodes of heterotopic ossification (HO) formation in muscles, tendons, and ligaments. FOP is caused by a missense mutation in the ACVR1 gene (activin A receptor type I), an important signaling receptor involved in endochondral ossification. The ACVR1R206H mutation induces increased downstream canonical SMAD-signaling and drives tissue-resident progenitor cells with osteogenic potential to participate in endochondral HO formation. In this article, we review aberrant ACVR1R206H signaling and the cells that give rise to HO in FOP. FOP mouse models and lineage tracing analyses have been used to provide strong evidence for tissue-resident mesenchymal cells as cellular contributors to HO. We assess how the underlying mutation in FOP disrupts muscle-specific dynamics during homeostasis and repair, with a focus on muscle-resident mesenchymal cells known as fibro-adipogenic progenitors (FAPs). Accumulating research points to FAPs as a prominent HO progenitor population, with ACVR1R206H FAPs not only aberrantly differentiating into chondro-osteogenic lineages but creating a permissive environment for bone formation at the expense of muscle regeneration. We will further discuss the emerging role of ACVR1R206H FAPs in muscle regeneration and therapeutic targeting of these cells to reduce HO formation in FOP.
Keywords: ACVR1 mutation; FOP; HO progenitor cells; fibro-adipogenic progenitors; heterotopic ossification; muscle regeneration; musculoskeletal disease.
Conflict of interest statement
The authors declare no conflicts of interest.
Figures
References
-
- Shore E.M., Xu M., Feldman G.J., Fenstermacher D.A., Cho T.J., Choi I.H., Connor J.M., Delai P., Glaser D.L., LeMerrer M., et al. A recurrent mutation in the BMP type I receptor ACVR1 causes inherited and sporadic fibrodysplasia ossificans progressiva. Nat. Genet. 2006;38:525–527. doi: 10.1038/ng1783. - DOI - PubMed
-
- Frederick S., Kaplan J.C.G., Meiqi Xu O., Towler W., Grunvald E., Kalunian K., Kallish S., Al Mukaddam M., Pignolo R.J., Eileen M. Shore An ACVR1R375P pathogenic variant in two families with mild fibrodysplasia ossificans progressiva. Am. J. Med. Genet. 2021;188:806–817. - PubMed
-
- Kaplan F.S., Xu M., Seemann P., Connor J.M., Glaser D.L., Carroll L., Delai P., Fastnacht-Urban E., Forman S.J., Gillessen-Kaesbach G., et al. Classic and atypical fibrodysplasia ossificans progressiva (FOP) phenotypes are caused by mutations in the bone morphogenetic protein (BMP) type I receptor ACVR1. Hum. Mutat. 2009;30:379–390. doi: 10.1002/humu.20868. - DOI - PMC - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
