

Exploring MiR-484 Regulation by Polyalthia longifolia: A Promising Biomarker and Therapeutic Target in Cervical Cancer through Integrated Bioinformatics and an In Vitro Analysis
- PMID: 38672263
- PMCID: PMC11047986
- DOI: 10.3390/biomedicines12040909
Exploring MiR-484 Regulation by Polyalthia longifolia: A Promising Biomarker and Therapeutic Target in Cervical Cancer through Integrated Bioinformatics and an In Vitro Analysis
Abstract
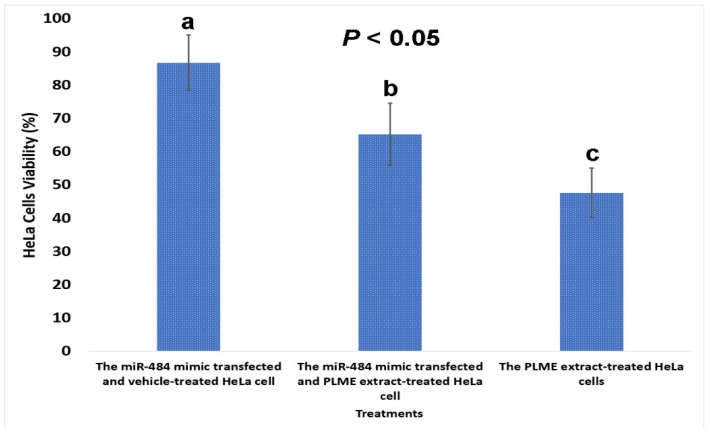
Background: MiR-484, implicated in various carcinomas, holds promise as a prognostic marker, yet its relevance to cervical cancer (CC) remains unclear. Our prior study demonstrated the Polyalthia longifolia downregulation of miR-484, inhibiting HeLa cells. This study investigates miR-484's potential as a biomarker and therapeutic target in CC through integrated bioinformatics and an in vitro analysis.
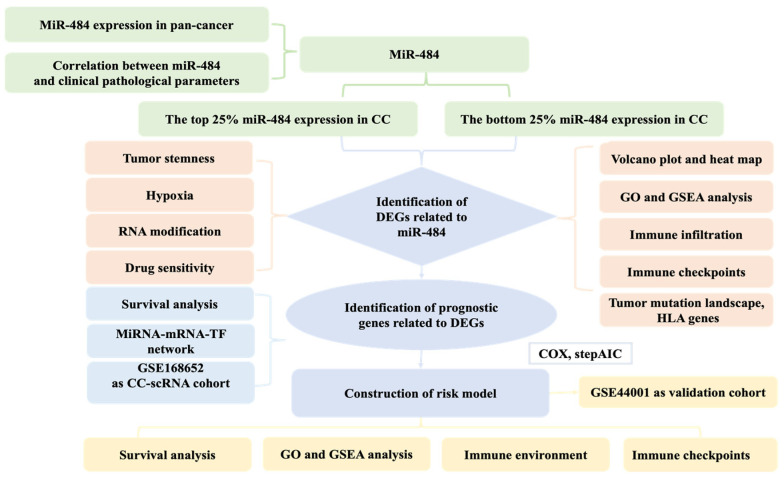
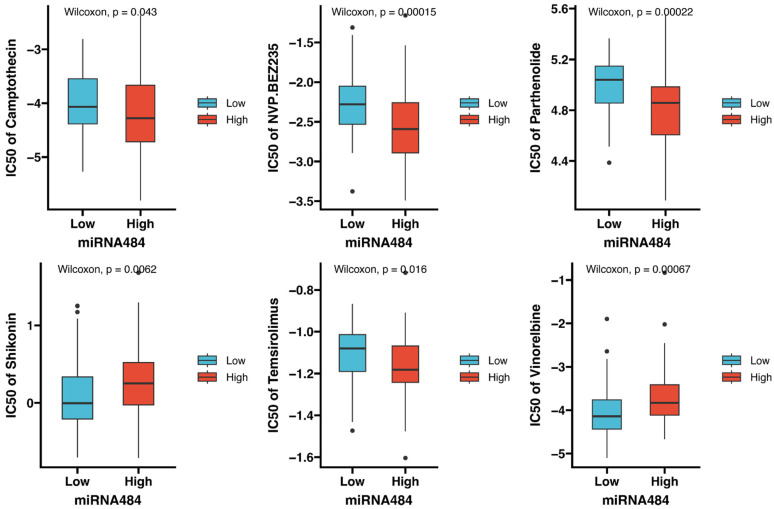
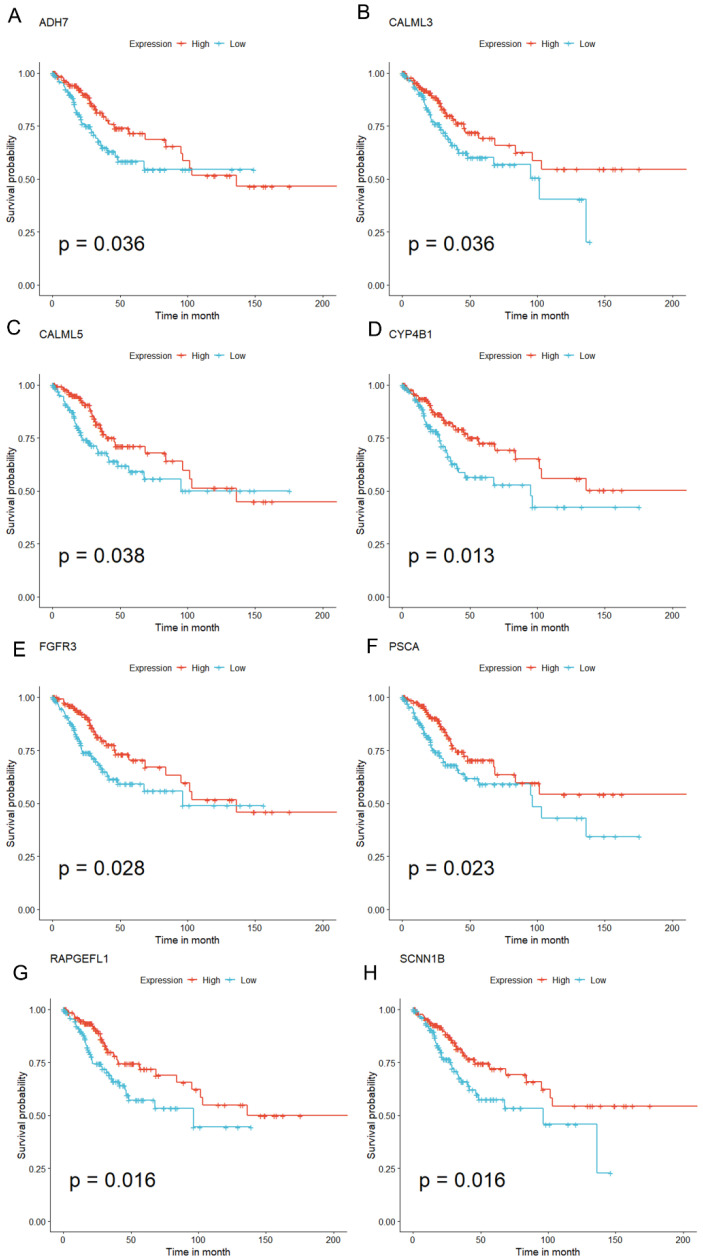
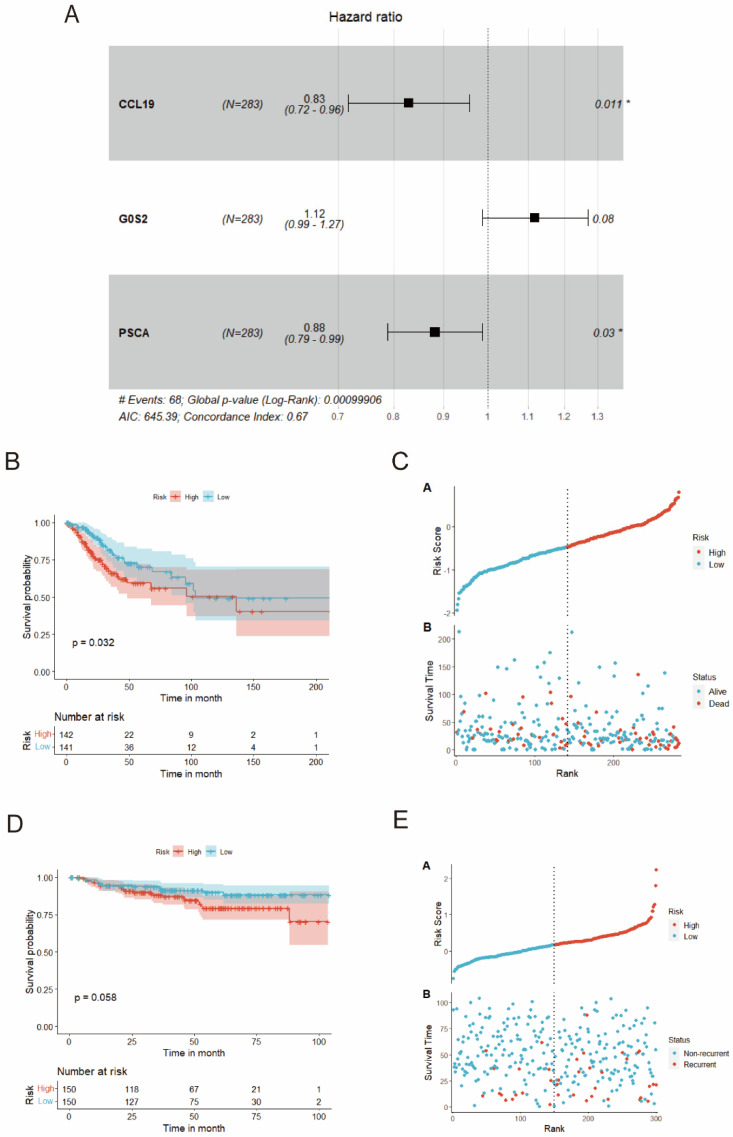
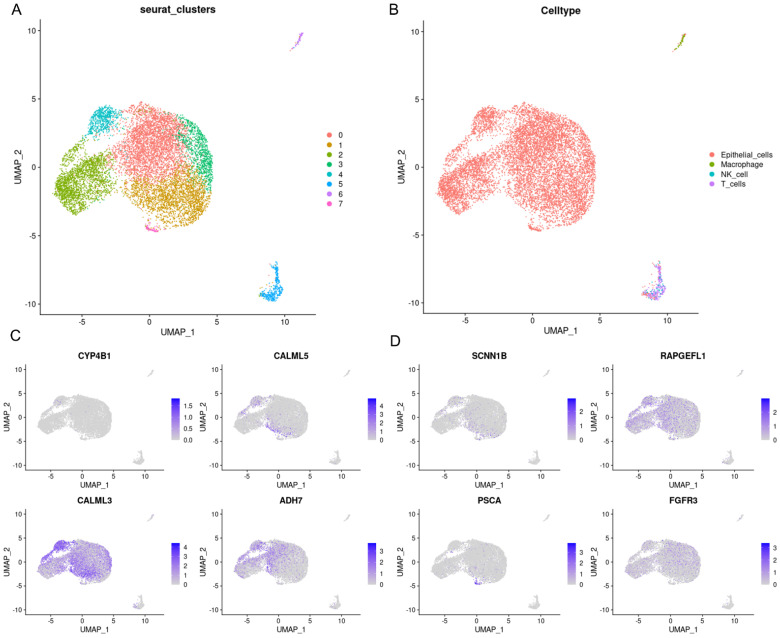
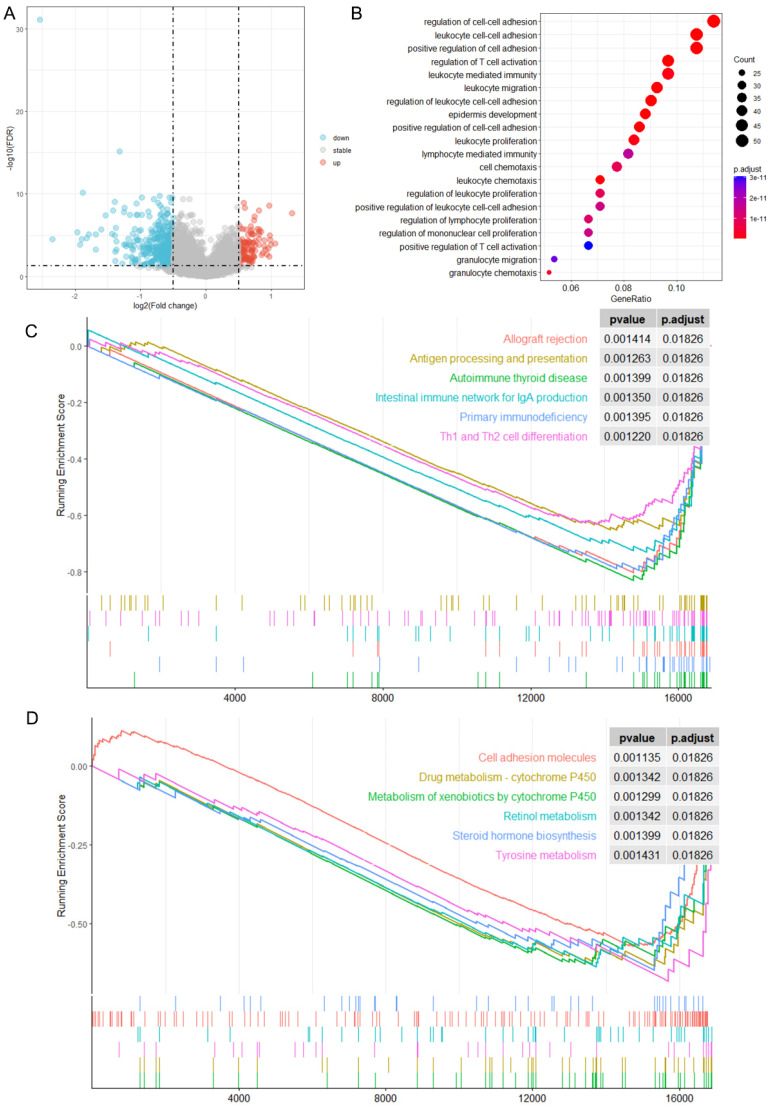
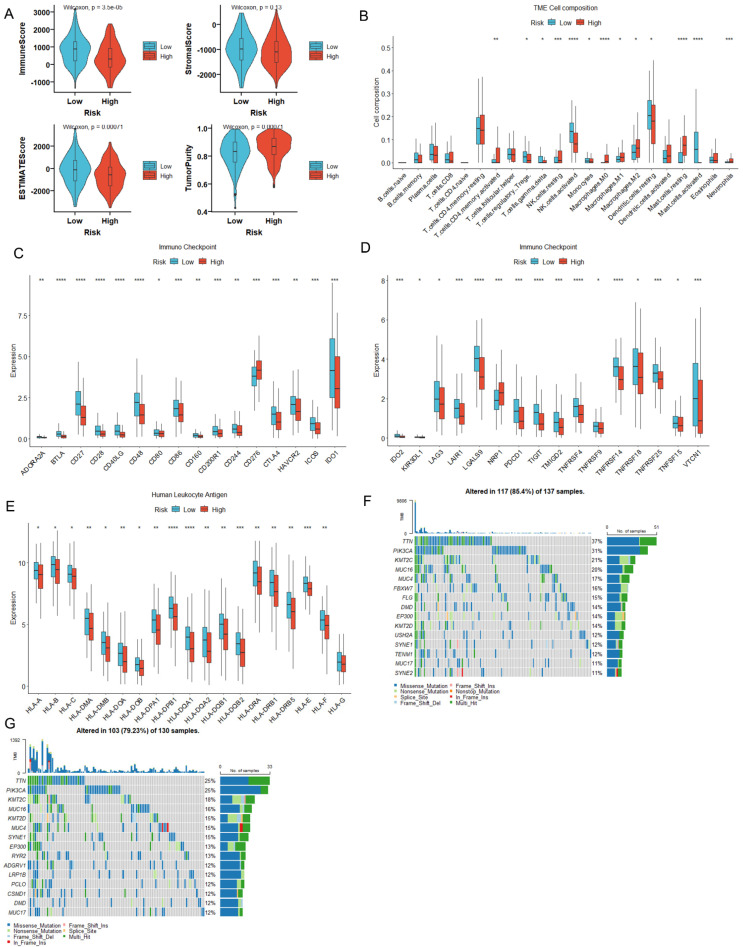
Methods: MiR-484 levels were analyzed across cancers, including CC, from The Cancer Genome Atlas. The limma R package identified differentially expressed genes (DEGs) between high- and low-miR-484 CC cohorts. We assessed biological functions, tumor microenvironment (TME), immunotherapy, stemness, hypoxia, RNA methylation, and chemosensitivity differences. Prognostic genes relevant to miR-484 were identified through Cox regression and Kaplan-Meier analyses, and a prognostic model was captured via multivariate Cox regression. Single-cell RNA sequencing determined cell populations related to prognostic genes. qRT-PCR validated key genes, and the miR-484 effect on CC proliferation was assessed via an MTT assay.
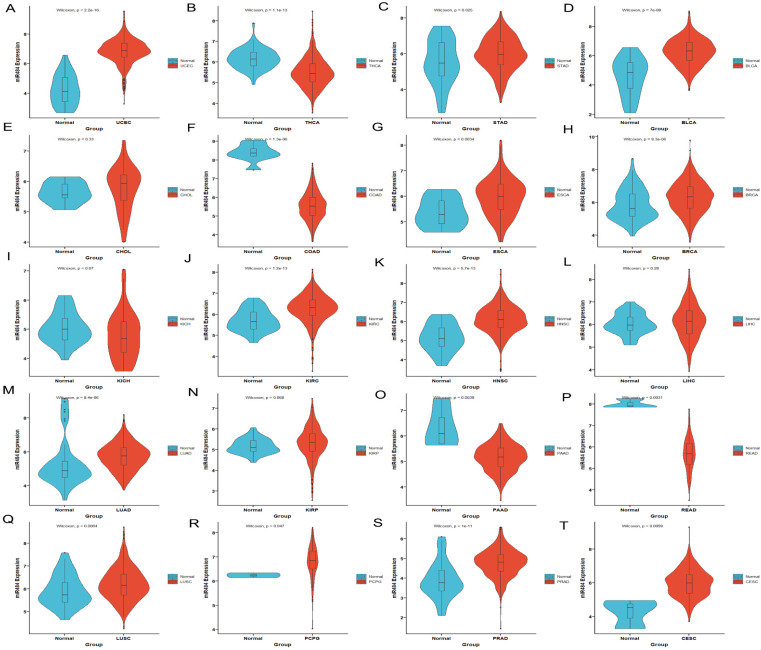
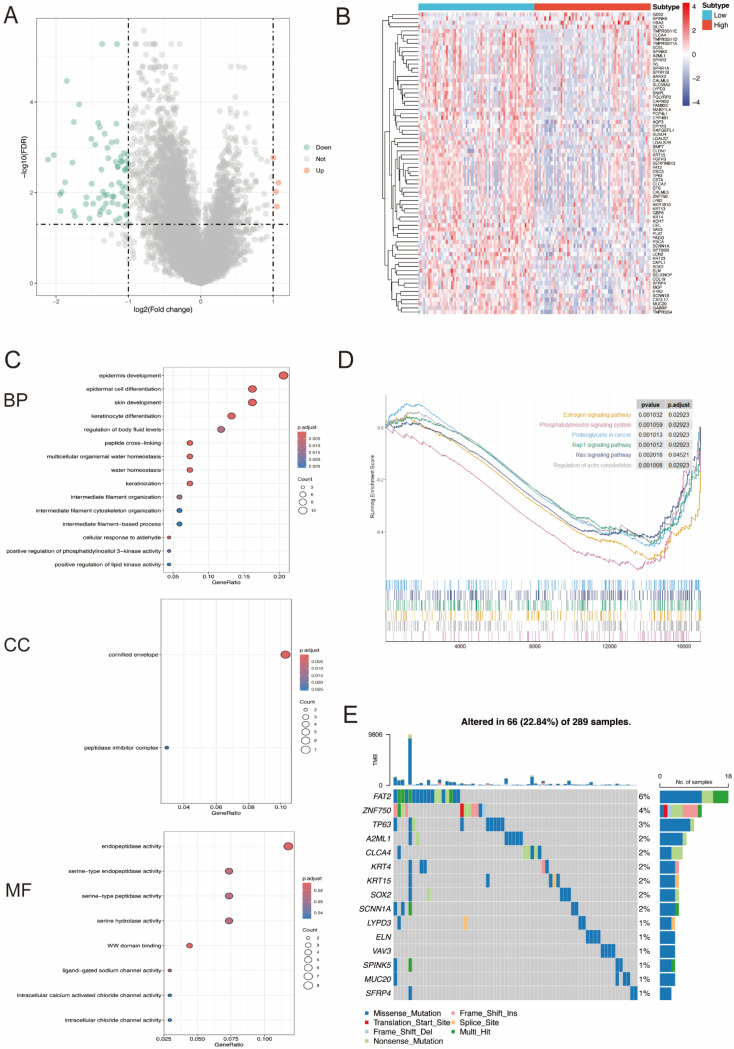
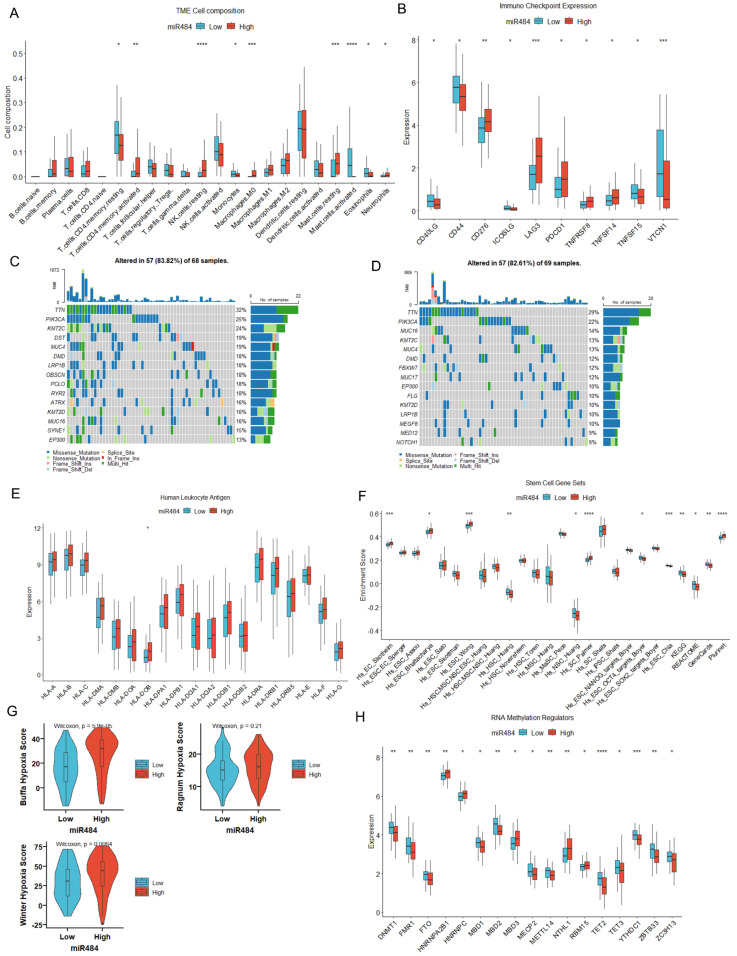
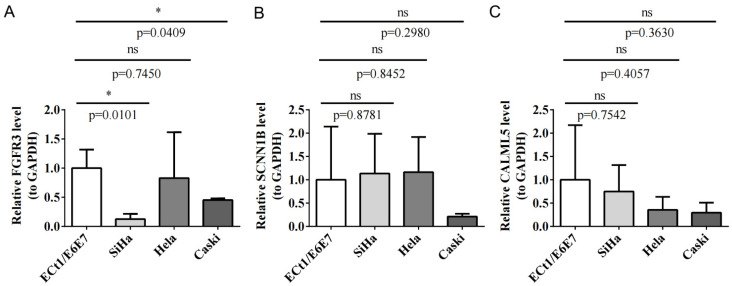
Results: MiR-484 was upregulated in most tumors, including CC, with DEGs enriched in skin development, PI3K signaling, and immune processes. High miR-484 expression correlated with specific immune cell infiltration, hypoxia, and drug sensitivity. Prognostic genes identified were predominantly epidermal and stratified patients with CC into risk groups, with the low-risk group showing enhanced survival and immunotherapeutic responses. qRT-PCR confirmed FGFR3 upregulation in CC cells, and an miR-484 mimic reversed the P. longifolia inhibitory effect on HeLa proliferation.
Conclusion: MiR-484 plays a crucial role in the CC progression and prognosis, suggesting its potential as a biomarker for targeted therapy.
Keywords: cervical cancer; immunotherapy; miR-484; prognostic genes; proliferation.
Conflict of interest statement
The authors declare no conflicts of interest.
Figures
References
-
- Doghish A.S., Ali M.A., Elyan S.S., Elrebehy M.A., Mohamed H.H., Mansour R.M., Elgohary A., Ghanem A., Faraag A.H.I., Abdelmaksoud N.M., et al. miRNAs role in cervical cancer pathogenesis and targeted therapy: Signaling pathways interplay. Pathol. Res. Pract. 2023;244:154386. doi: 10.1016/j.prp.2023.154386. - DOI - PubMed
LinkOut - more resources
Full Text Sources
