

Alcohol-Associated Liver Disease Outcomes: Critical Mechanisms of Liver Injury Progression
- PMID: 38672422
- PMCID: PMC11048648
- DOI: 10.3390/biom14040404
Alcohol-Associated Liver Disease Outcomes: Critical Mechanisms of Liver Injury Progression
Abstract
Alcohol-associated liver disease (ALD) is a substantial cause of morbidity and mortality worldwide and represents a spectrum of liver injury beginning with hepatic steatosis (fatty liver) progressing to inflammation and culminating in cirrhosis. Multiple factors contribute to ALD progression and disease severity. Here, we overview several crucial mechanisms related to ALD end-stage outcome development, such as epigenetic changes, cell death, hemolysis, hepatic stellate cells activation, and hepatic fatty acid binding protein 4. Additionally, in this review, we also present two clinically relevant models using human precision-cut liver slices and hepatic organoids to examine ALD pathogenesis and progression.
Keywords: MetAld; alcohol-associated liver disease; cell death; epigenetics; fatty acid binding protein 4; fibrosis; hemolysis; hepatic stellate cells; hepatocellular carcinoma; models.
Conflict of interest statement
The authors declare no conflicts of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
Figures
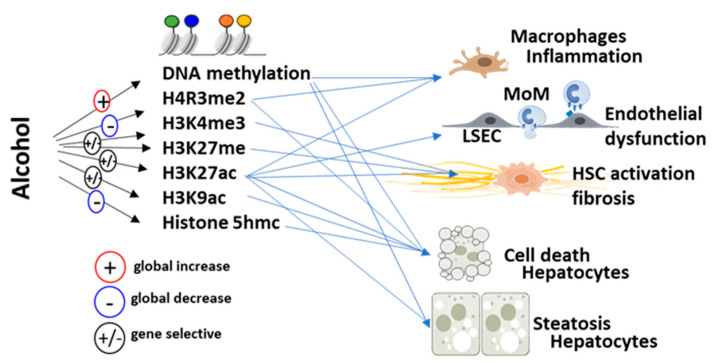
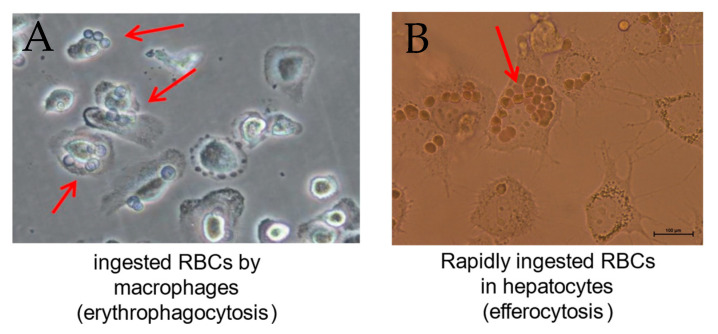
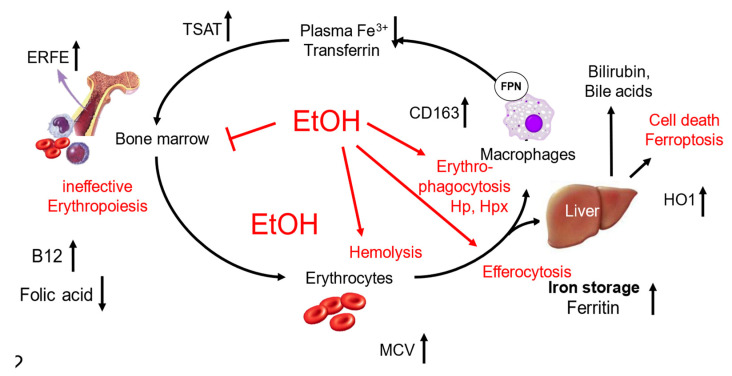
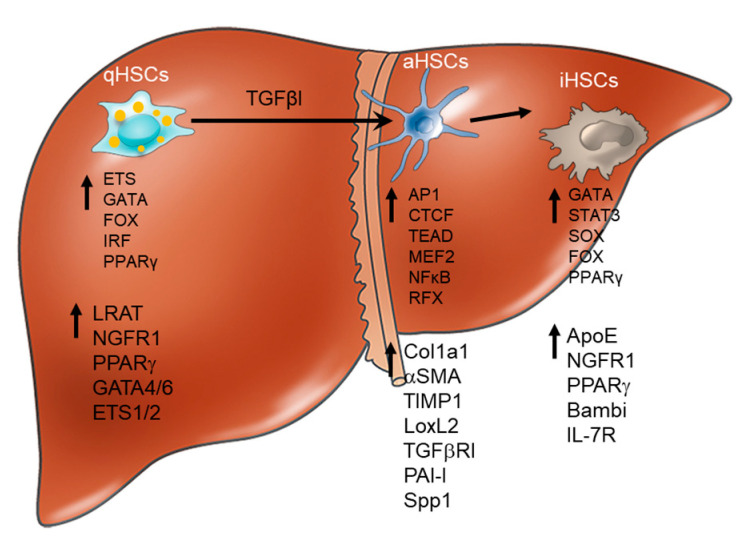
 denotes up-regulation.
denotes up-regulation.References
Publication types
MeSH terms
Grants and funding
- I01 BX006064/BX/BLRD VA/United States
- P42 ES010337/ES/NIEHS NIH HHS/United States
- R21 AA029241/AA/NIAAA NIH HHS/United States
- U01 AA029019/AA/NIAAA NIH HHS/United States
- R01 DK091183/DK/NIDDK NIH HHS/United States
- R01 AA028550/AA/NIAAA NIH HHS/United States
- P50 AA030407/AA/NIAAA NIH HHS/United States
- R01 AA026723/AA/NIAAA NIH HHS/United States
- R01 AA020744/AA/NIAAA NIH HHS/United States
- R01 AA027189/AA/NIAAA NIH HHS/United States
- I01 BX004053/BX/BLRD VA/United States
- R01 AA011576/AA/NIAAA NIH HHS/United States
- P30 DK120515/DK/NIDDK NIH HHS/United States
- R01 AA027586/AA/NIAAA NIH HHS/United States
- R01 DK133930/DK/NIDDK NIH HHS/United States
- R01 AA017729/AA/NIAAA NIH HHS/United States
- R01 DK111866/DK/NIDDK NIH HHS/United States
- R01 DK137061/DK/NIDDK NIH HHS/United States
- R01 DK099205/DK/NIDDK NIH HHS/United States
- P50 AA011999/AA/NIAAA NIH HHS/United States
- R01 DK135817/DK/NIDDK NIH HHS/United States
- R01 DK101737/DK/NIDDK NIH HHS/United States
- R44 DK115242/DK/NIDDK NIH HHS/United States
LinkOut - more resources
Full Text Sources
Research Materials
