

Coagulation Protease-Driven Cancer Immune Evasion: Potential Targets for Cancer Immunotherapy
- PMID: 38672649
- PMCID: PMC11048528
- DOI: 10.3390/cancers16081568
Coagulation Protease-Driven Cancer Immune Evasion: Potential Targets for Cancer Immunotherapy
Abstract
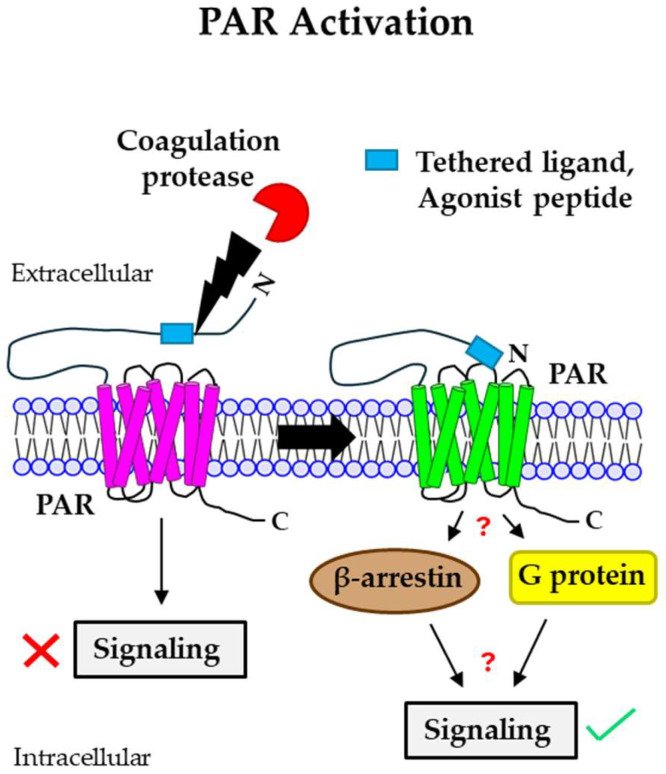
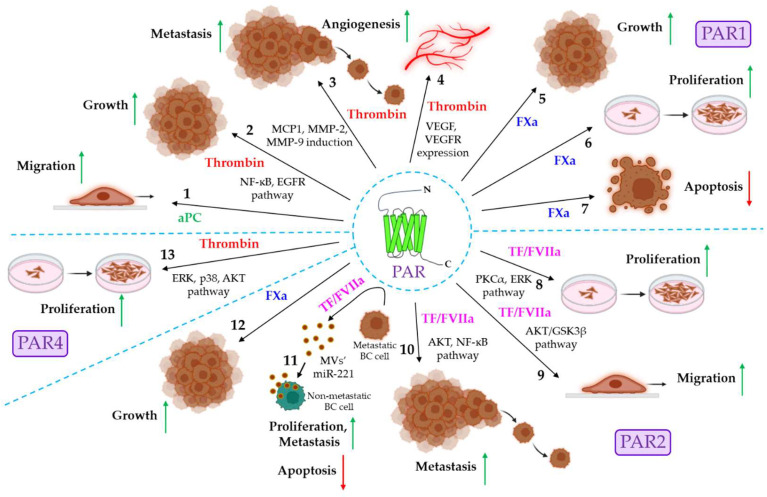
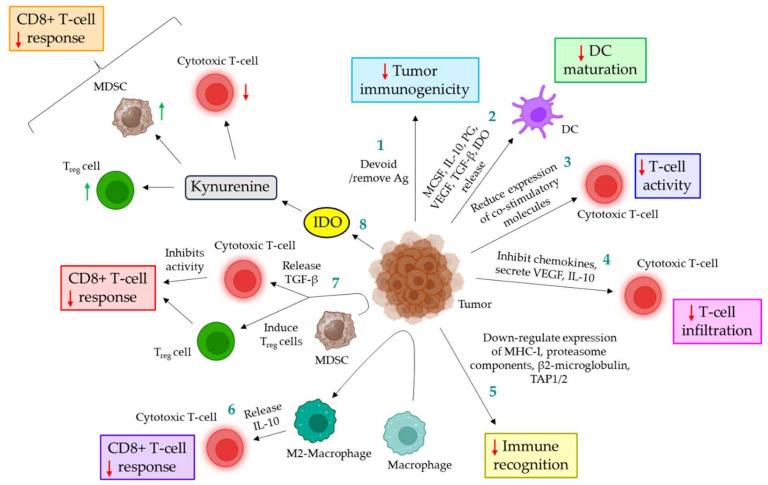
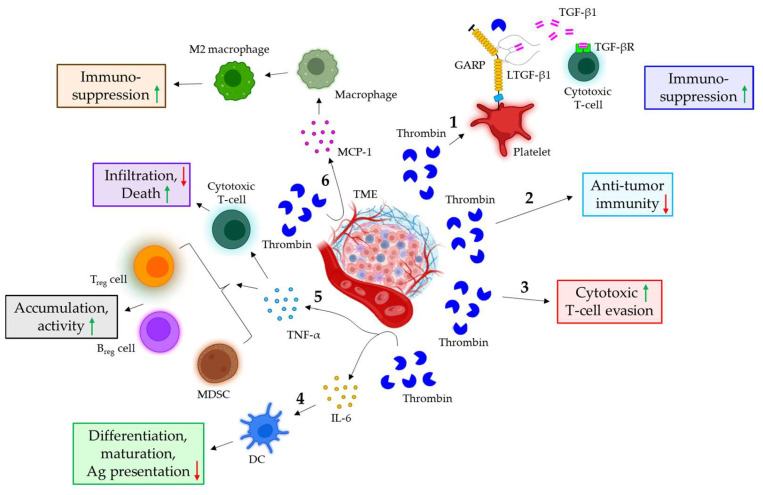
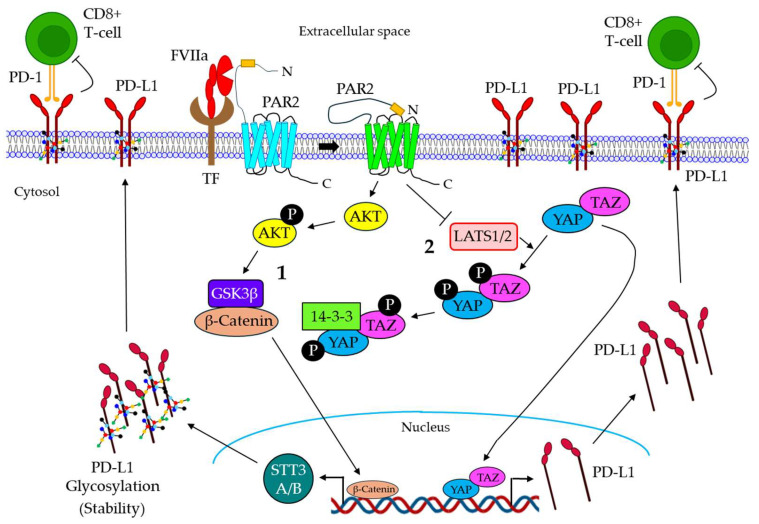
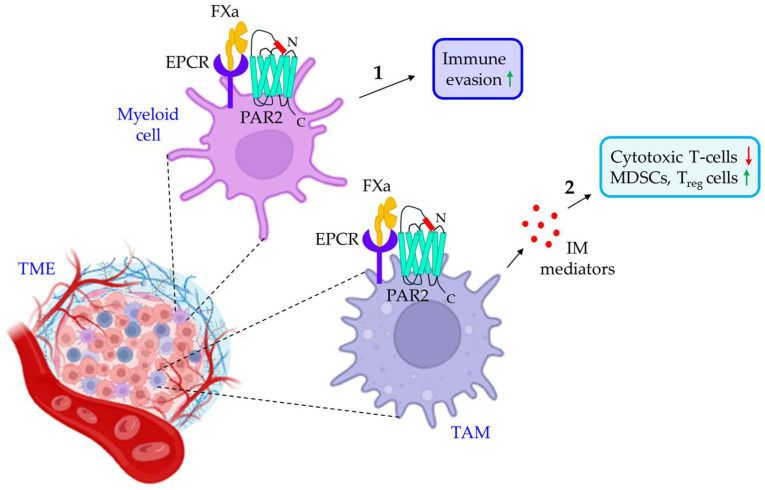
Blood coagulation and cancer are intrinsically connected, hypercoagulation-associated thrombotic complications are commonly observed in certain types of cancer, often leading to decreased survival in cancer patients. Apart from the common role in coagulation, coagulation proteases often trigger intracellular signaling in various cancers via the activation of a G protein-coupled receptor superfamily protease: protease-activated receptors (PARs). Although the role of PARs is well-established in the development and progression of certain types of cancer, their impact on cancer immune response is only just emerging. The present review highlights how coagulation protease-driven PAR signaling plays a key role in modulating innate and adaptive immune responses. This is followed by a detailed discussion on the contribution of coagulation protease-induced signaling in cancer immune evasion, thereby supporting the growth and development of certain tumors. A special section of the review demonstrates the role of coagulation proteases, thrombin, factor VIIa, and factor Xa in cancer immune evasion. Targeting coagulation protease-induced signaling might be a potential therapeutic strategy to boost the immune surveillance mechanism of a host fighting against cancer, thereby augmenting the clinical consequences of targeted immunotherapeutic regimens.
Keywords: biomarker; blood coagulation; cancer; clinical trial; coagulation protease; immune evasion; immunotherapy; protease-activated receptor.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
Similar articles
-
Mechanistic coupling of protease signaling and initiation of coagulation by tissue factor.Proc Natl Acad Sci U S A. 2001 Jul 3;98(14):7742-7. doi: 10.1073/pnas.141126698. Proc Natl Acad Sci U S A. 2001. PMID: 11438726 Free PMC article.
-
Protease-activated receptor signalling by coagulation proteases in endothelial cells.Thromb Haemost. 2014 Nov;112(5):876-82. doi: 10.1160/TH14-02-0167. Epub 2014 Jul 3. Thromb Haemost. 2014. PMID: 24990498 Free PMC article. Review.
-
Homeostatic effects of coagulation protease-dependent signaling and protease activated receptors.J Thromb Haemost. 2017 Jul;15(7):1273-1284. doi: 10.1111/jth.13721. J Thromb Haemost. 2017. PMID: 28671351 Review.
-
Coagulation signaling and cancer immunotherapy.Thromb Res. 2020 Jul;191 Suppl 1:S106-S111. doi: 10.1016/S0049-3848(20)30406-0. Thromb Res. 2020. PMID: 32736766
-
Coagulation-independent effects of thrombin and Factor Xa: role of protease-activated receptors in pulmonary hypertension.Cardiovasc Res. 2022 Dec 29;118(16):3225-3238. doi: 10.1093/cvr/cvac004. Cardiovasc Res. 2022. PMID: 35104324
Cited by
-
The tumor coagulome as a potential biological determinant of postsurgical recurrence of oral squamous cell carcinoma.Front Oral Health. 2025 Feb 24;6:1554739. doi: 10.3389/froh.2025.1554739. eCollection 2025. Front Oral Health. 2025. PMID: 40065838 Free PMC article.
-
O-GlcNAcylation: Crosstalk between Hemostasis, Inflammation, and Cancer.Int J Mol Sci. 2024 Sep 13;25(18):9896. doi: 10.3390/ijms25189896. Int J Mol Sci. 2024. PMID: 39337387 Free PMC article. Review.
-
MicroRNAs in lung cancer: their role in tumor progression, biomarkers, diagnostic, prognostic, and therapeutic relevance.Discov Oncol. 2025 Mar 11;16(1):293. doi: 10.1007/s12672-025-02054-9. Discov Oncol. 2025. PMID: 40067551 Free PMC article. Review.
-
Machine Learning-Based Integration of Single-Cell and Bulk Transcriptome Reveals Coagulation Signature and Phenotypic Heterogeneity in Hepatocellular Carcinoma.IET Syst Biol. 2025 Jan-Dec;19(1):e70033. doi: 10.1049/syb2.70033. IET Syst Biol. 2025. PMID: 40820328 Free PMC article.
References
Publication types
LinkOut - more resources
Full Text Sources
Miscellaneous
