

Genome Analysis of Epsilon CrAss-like Phages
- PMID: 38675856
- PMCID: PMC11054128
- DOI: 10.3390/v16040513
Genome Analysis of Epsilon CrAss-like Phages
Abstract
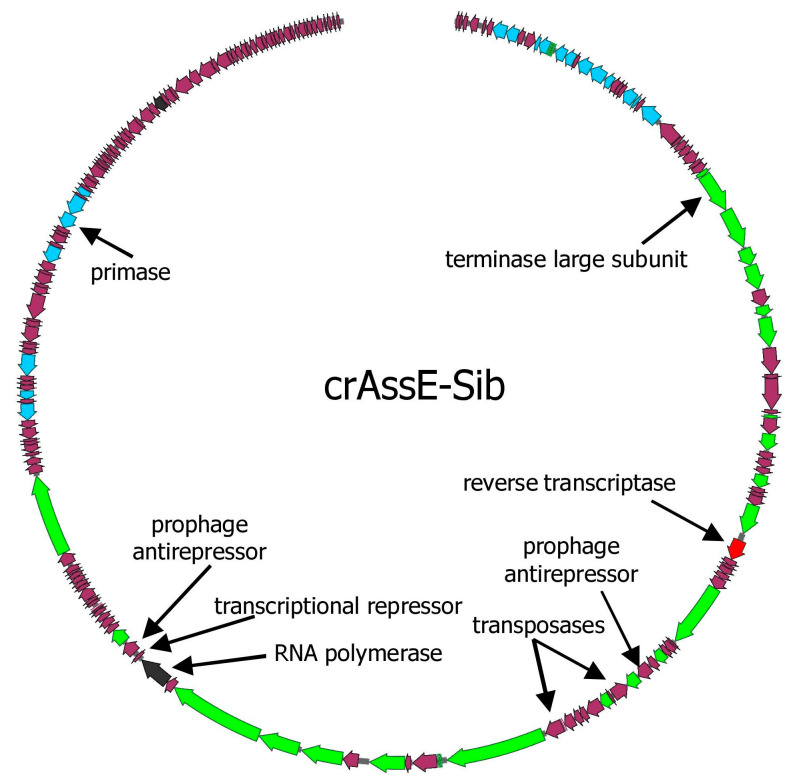
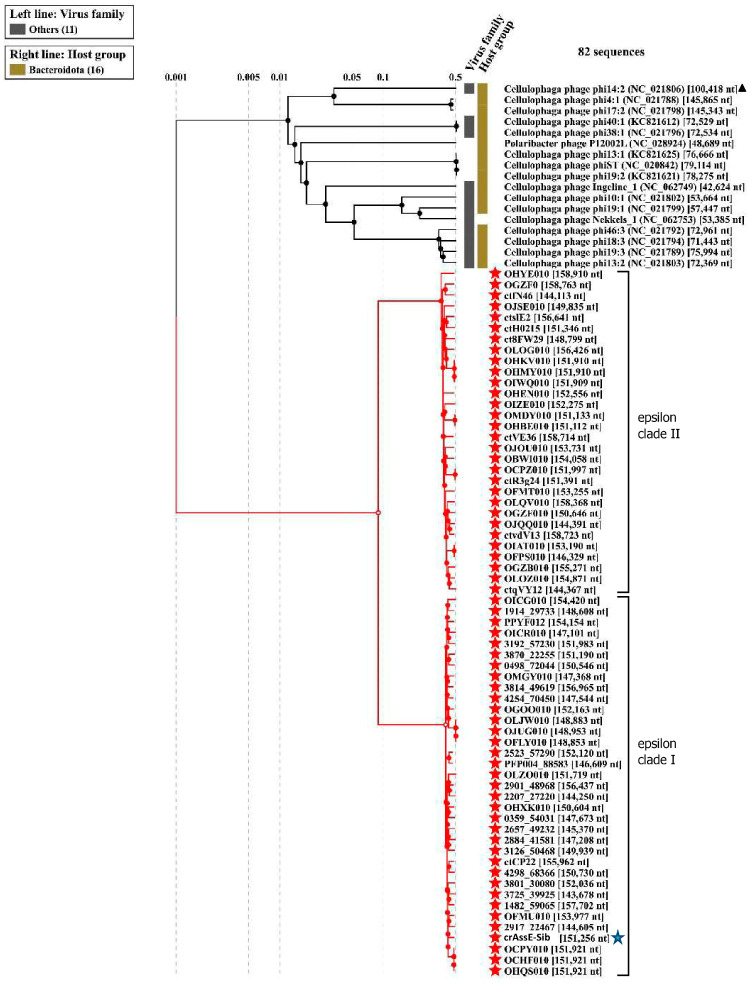
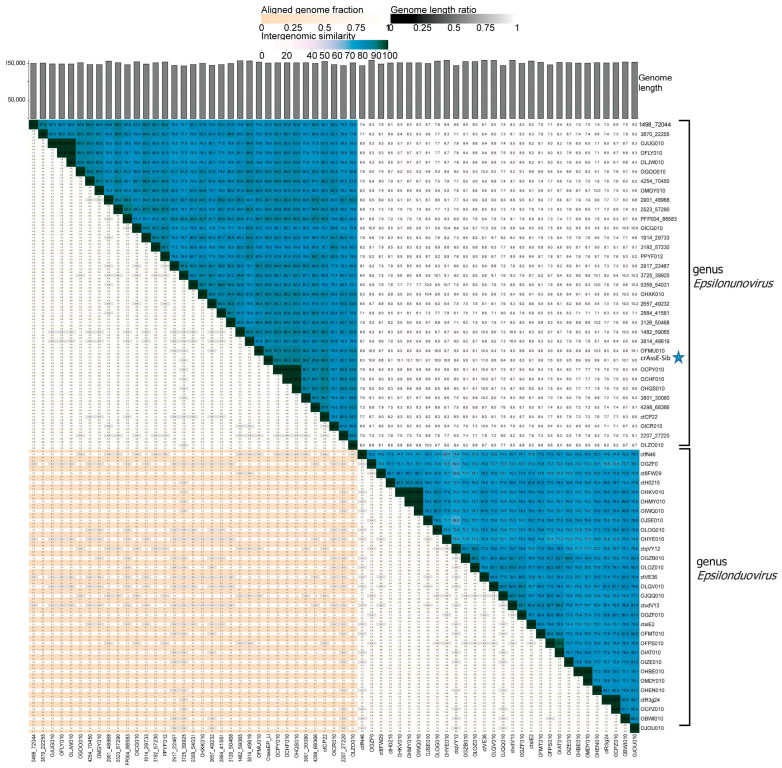
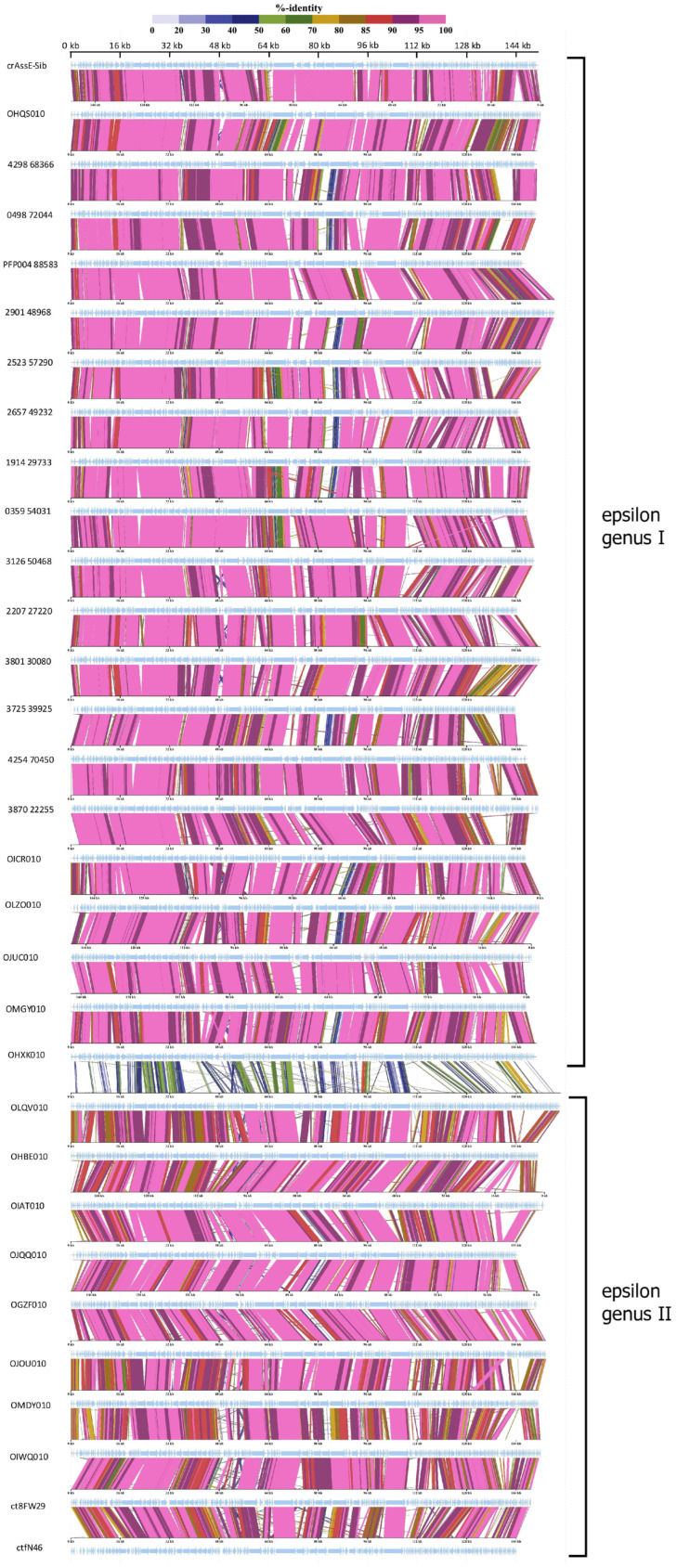
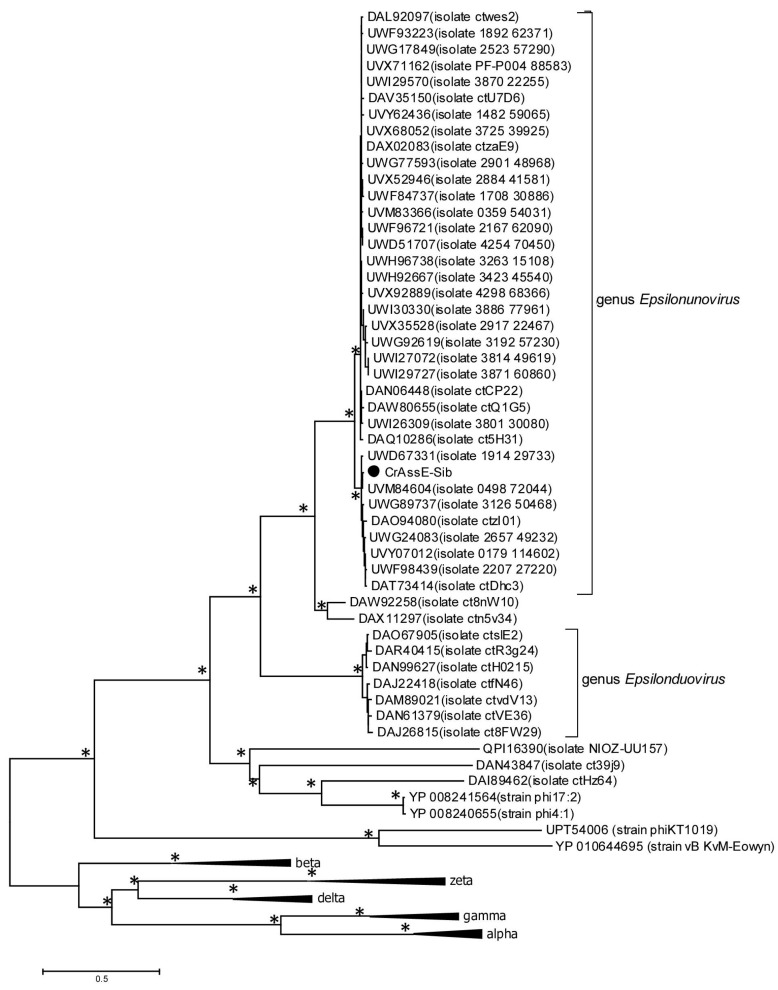
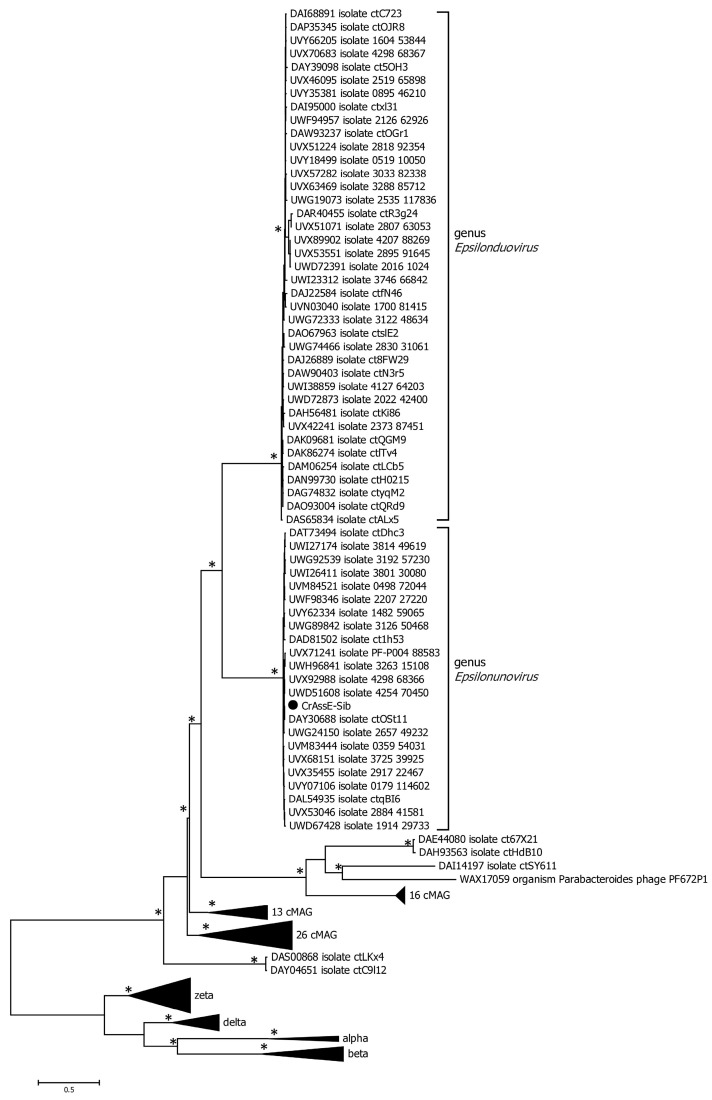
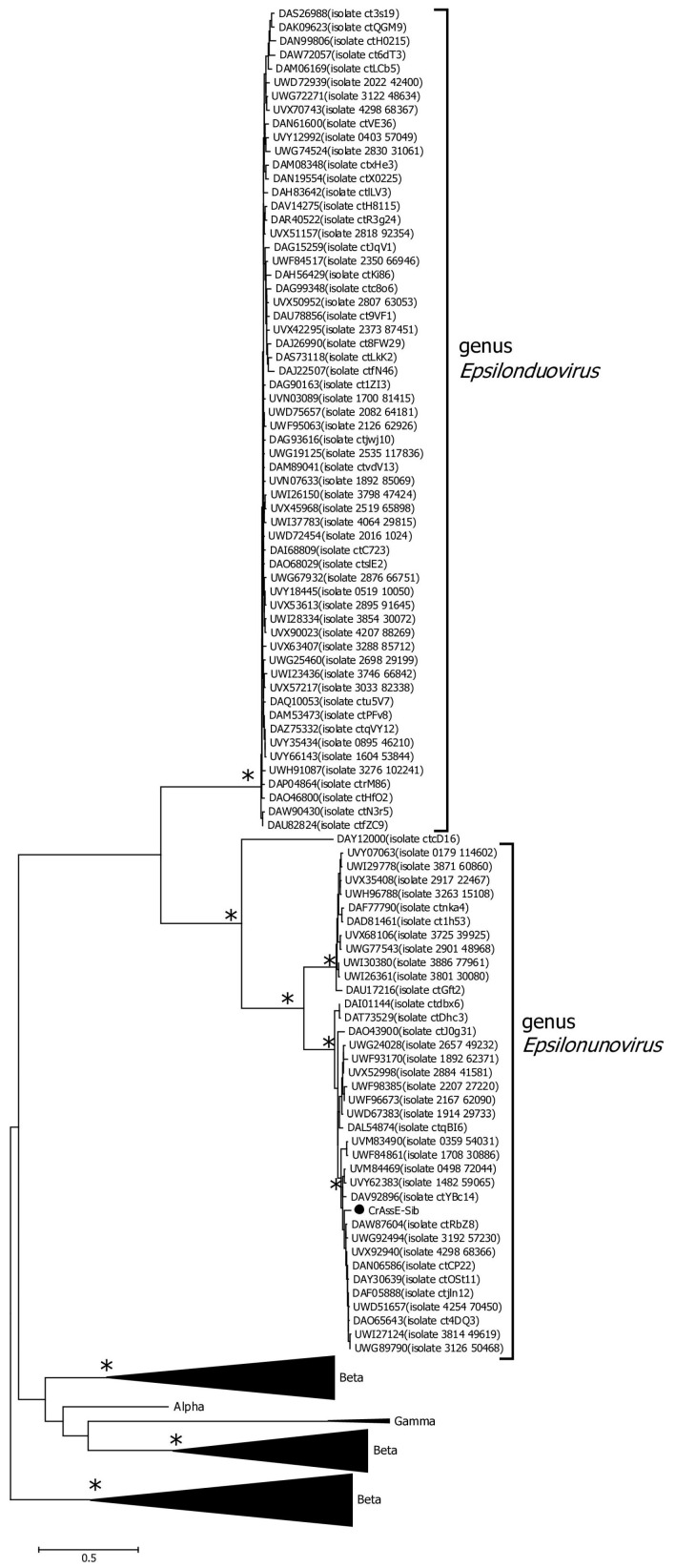
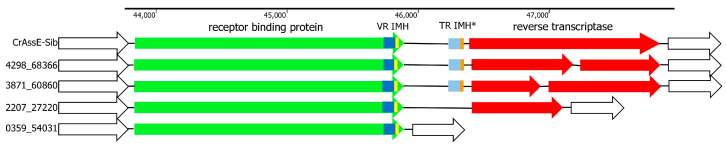
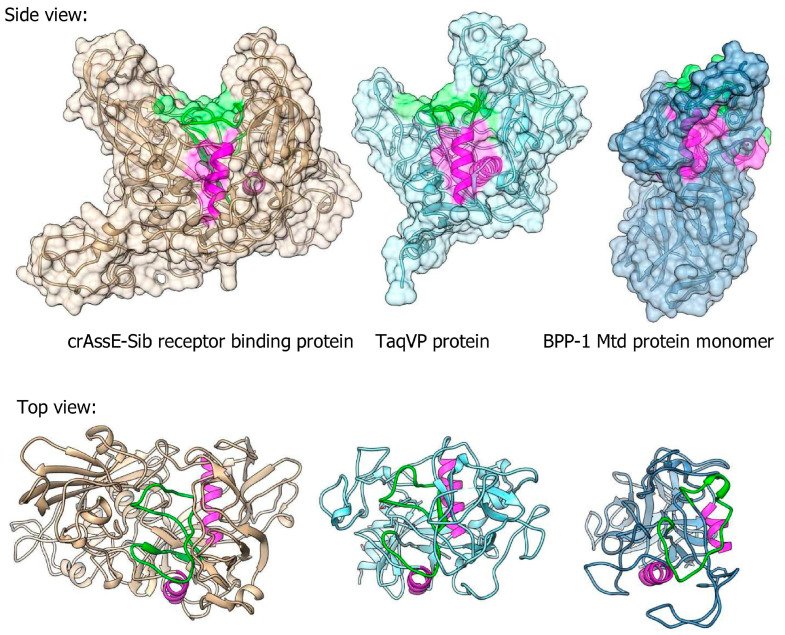
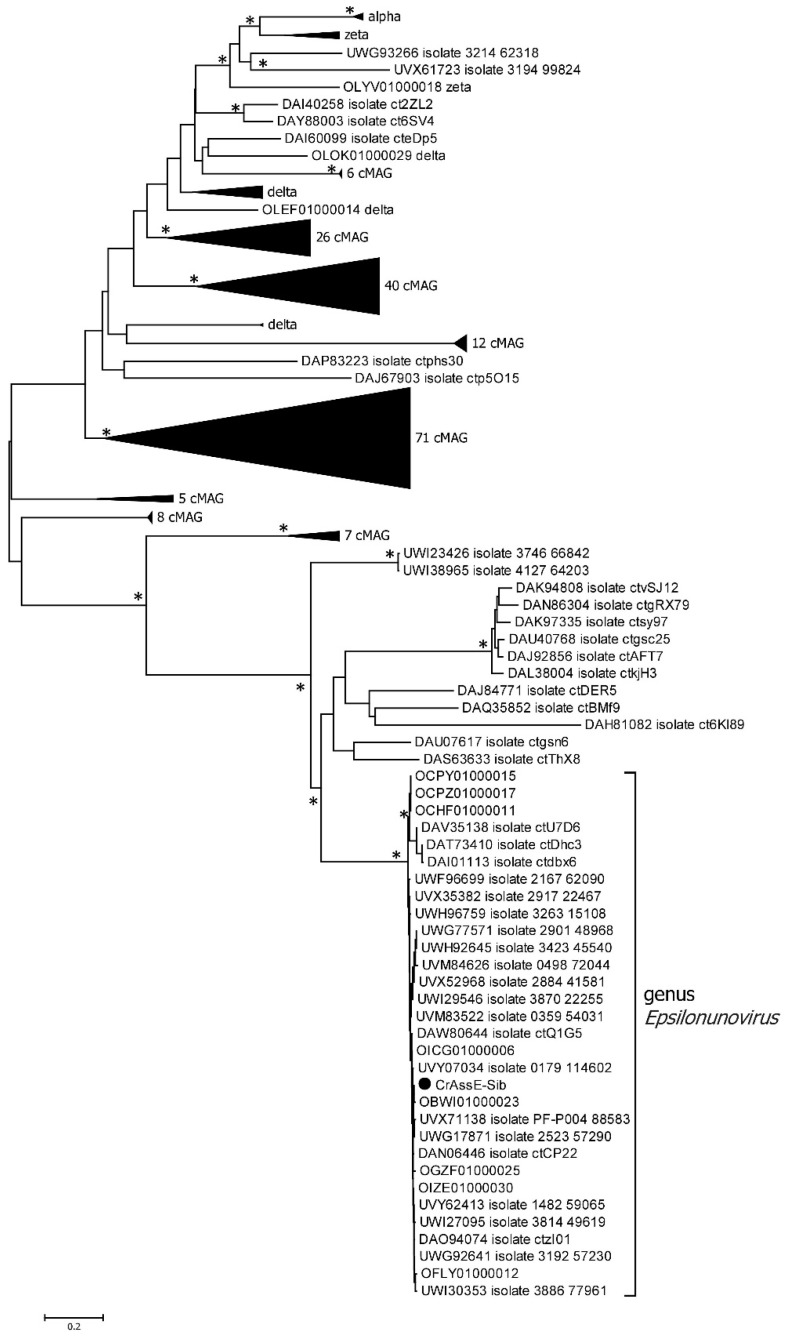
CrAss-like phages play an important role in maintaining ecological balance in the human intestinal microbiome. However, their genetic diversity and lifestyle are still insufficiently studied. In this study, a novel CrAssE-Sib phage genome belonging to the epsilon crAss-like phage genomes was found. Comparative analysis indicated that epsilon crAss-like phages are divided into two putative genera, which were proposed to be named Epsilonunovirus and Epsilonduovirus; CrAssE-Sib belongs to the former. The crAssE-Sib genome contains a diversity-generating retroelement (DGR) cassette with all essential elements, including the reverse transcriptase (RT) and receptor binding protein (RBP) genes. However, this RT contains the GxxxSP motif in its fourth domain instead of the usual GxxxSQ motif found in all known phage and bacterial DGRs. RBP encoded by CrAssE-Sib and other Epsilonunoviruses has an unusual structure, and no similar phage proteins were found. In addition, crAssE-Sib and other Epsilonunoviruses encode conserved prophage repressor and anti-repressors that could be involved in lysogenic-to-lytic cycle switches. Notably, DNA primase sequences of epsilon crAss-like phages are not included in the monophyletic group formed by the DNA primases of all other crAss-like phages. Therefore, epsilon crAss-like phage substantially differ from other crAss-like phages, indicating the need to classify these phages into a separate family.
Keywords: crAss-like phages; diversity-generating retroelements; genome; repressors/anti-repressors; reverse transcriptase; virus taxonomy.
Conflict of interest statement
All co-authors have seen and agree with the contents of the manuscript and the order of authors, and there is no financial interest to report. All co-authors declare that they have no conflicts of interest.
Figures

References
-
- Guerin E., Shkoporov A., Stockdale S.R., Clooney A.G., Ryan F.J., Sutton T.D.S., Draper L.A., Gonzalez-Tortuero E., Ross R.P., Hill C. Biology and taxonomy of crAss-like Bacteriophages, the most abundant virus in the human gut. Cell Host Microbe. 2018;24:653–664. doi: 10.1016/j.chom.2018.10.002. - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
