

Molecular mechanisms and therapeutic strategies for neuromuscular diseases
- PMID: 38678519
- PMCID: PMC11056344
- DOI: 10.1007/s00018-024-05229-9
Molecular mechanisms and therapeutic strategies for neuromuscular diseases
Erratum in
-
Correction: Molecular mechanisms and therapeutic strategies for neuromuscular diseases.Cell Mol Life Sci. 2024 Jul 26;81(1):308. doi: 10.1007/s00018-024-05305-0. Cell Mol Life Sci. 2024. PMID: 39060715 Free PMC article. No abstract available.
Abstract
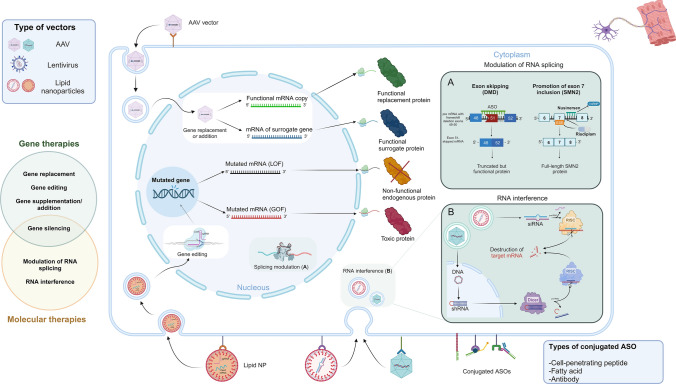
Neuromuscular diseases encompass a heterogeneous array of disorders characterized by varying onset ages, clinical presentations, severity, and progression. While these conditions can stem from acquired or inherited causes, this review specifically focuses on disorders arising from genetic abnormalities, excluding metabolic conditions. The pathogenic defect may primarily affect the anterior horn cells, the axonal or myelin component of peripheral nerves, the neuromuscular junction, or skeletal and/or cardiac muscles. While inherited neuromuscular disorders have been historically deemed not treatable, the advent of gene-based and molecular therapies is reshaping the treatment landscape for this group of condition. With the caveat that many products still fail to translate the positive results obtained in pre-clinical models to humans, both the technological development (e.g., implementation of tissue-specific vectors) as well as advances on the knowledge of pathogenetic mechanisms form a collective foundation for potentially curative approaches to these debilitating conditions. This review delineates the current panorama of therapies targeting the most prevalent forms of inherited neuromuscular diseases, emphasizing approved treatments and those already undergoing human testing, offering insights into the state-of-the-art interventions.
Keywords: Motor neuron disease; Myopathy; Neuromuscular junction; Neuropathy; Therapy.
© 2024. The Author(s).
Conflict of interest statement
Financial interests: Authors AAZ, YMF and AB declare they have no financial interests. Author SCP has received speaker and consultant honoraria from Company Argenx, Alia Therapeutics, LSC Lifescience. Author SCP has served on advisory boards for Company Argenx, Wave Therapeutics, Esperare.
Figures
References
-
- Mercuri E, Muntoni F, Baranello G et al (2021) Onasemnogene abeparvovec gene therapy for symptomatic infantile-onset spinal muscular atrophy type 1 (STR1VE-EU): an open-label, single-arm, multicentre, phase 3 trial. Lancet Neurol 20(10):832–841. 10.1016/s1474-4422(21)00251-9 10.1016/s1474-4422(21)00251-9 - DOI - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Medical
