

Mitochondrial Ca2+ Uniporter-Dependent Energetic Dysfunction Drives Hypertrophy in Heart Failure
- PMID: 38680963
- PMCID: PMC11055214
- DOI: 10.1016/j.jacbts.2024.01.007
Mitochondrial Ca2+ Uniporter-Dependent Energetic Dysfunction Drives Hypertrophy in Heart Failure
Abstract
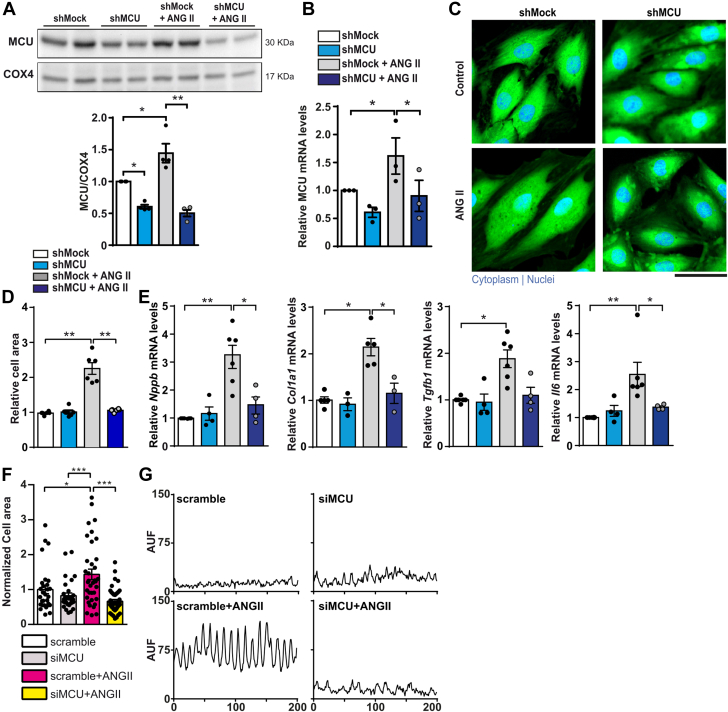
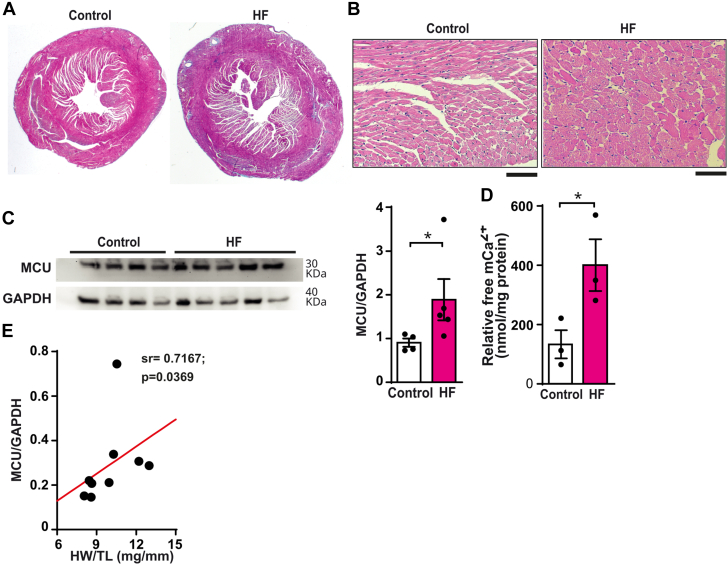
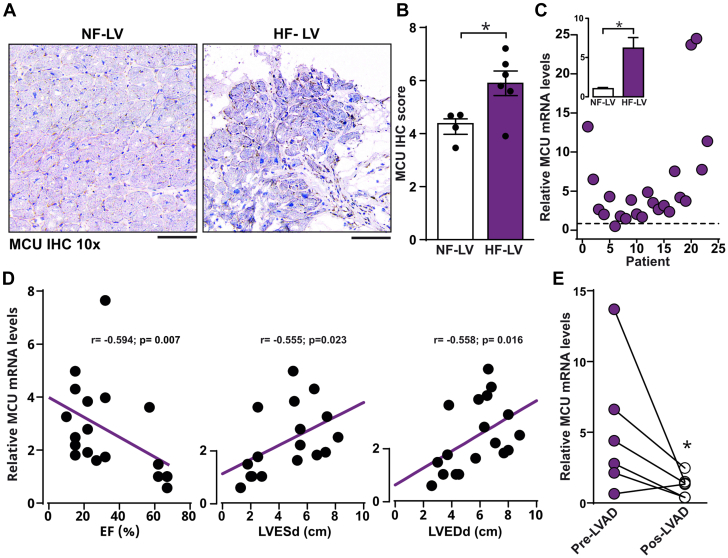
The role of the mitochondrial calcium uniporter (MCU) in energy dysfunction and hypertrophy in heart failure (HF) remains unknown. In angiotensin II (ANGII)-induced hypertrophic cardiac cells we have shown that hypertrophic cells overexpress MCU and present bioenergetic dysfunction. However, by silencing MCU, cell hypertrophy and mitochondrial dysfunction are prevented by blocking mitochondrial calcium overload, increase mitochondrial reactive oxygen species, and activation of nuclear factor kappa B-dependent hypertrophic and proinflammatory signaling. Moreover, we identified a calcium/calmodulin-independent protein kinase II/cyclic adenosine monophosphate response element-binding protein signaling modulating MCU upregulation by ANGII. Additionally, we found upregulation of MCU in ANGII-induced left ventricular HF in mice, and in the LV of HF patients, which was correlated with pathological remodeling. Following left ventricular assist device implantation, MCU expression decreased, suggesting tissue plasticity to modulate MCU expression.
Keywords: heart failure; mitochondrial calcium overload; mitochondrial calcium uniporter; mitochondrial dysfunction; pathological remodeling; reactive oxygen species.
© 2024 The Authors.
Conflict of interest statement
This work was partially supported by the CONACYT Grants 256577, 258197, Fronteras de la Ciencia Grant (0682). The authors have reported that they have no relationships relevant to the contents of this paper to disclose.
Figures






References
-
- Bers D.M. Cardiac excitation–contraction coupling. Nature. 2002;415(6868):198–205. - PubMed
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
