

Incomplete-penetrant hypertrophic cardiomyopathy MYH7 G256E mutation causes hypercontractility and elevated mitochondrial respiration
- PMID: 38683993
- PMCID: PMC11087781
- DOI: 10.1073/pnas.2318413121
Incomplete-penetrant hypertrophic cardiomyopathy MYH7 G256E mutation causes hypercontractility and elevated mitochondrial respiration
Erratum in
-
Correction for Lee et al., Incomplete-penetrant hypertrophic cardiomyopathy MYH7 G256E mutation causes hypercontractility and elevated mitochondrial respiration.Proc Natl Acad Sci U S A. 2024 Jun 25;121(26):e2410753121. doi: 10.1073/pnas.2410753121. Epub 2024 Jun 17. Proc Natl Acad Sci U S A. 2024. PMID: 38885393 Free PMC article. No abstract available.
Abstract
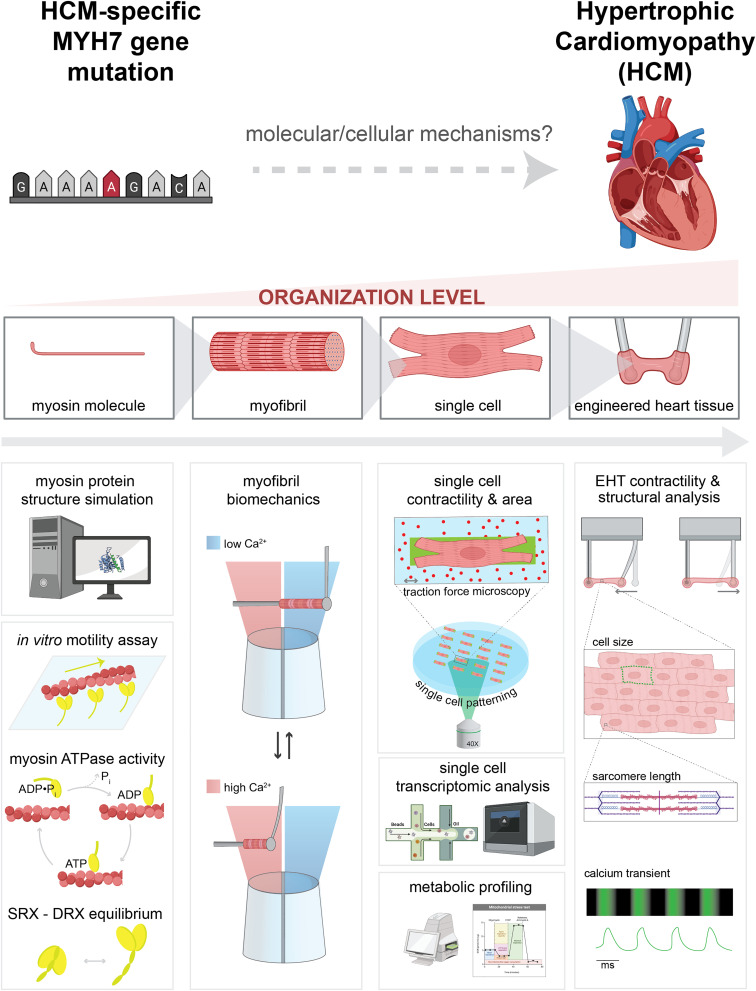
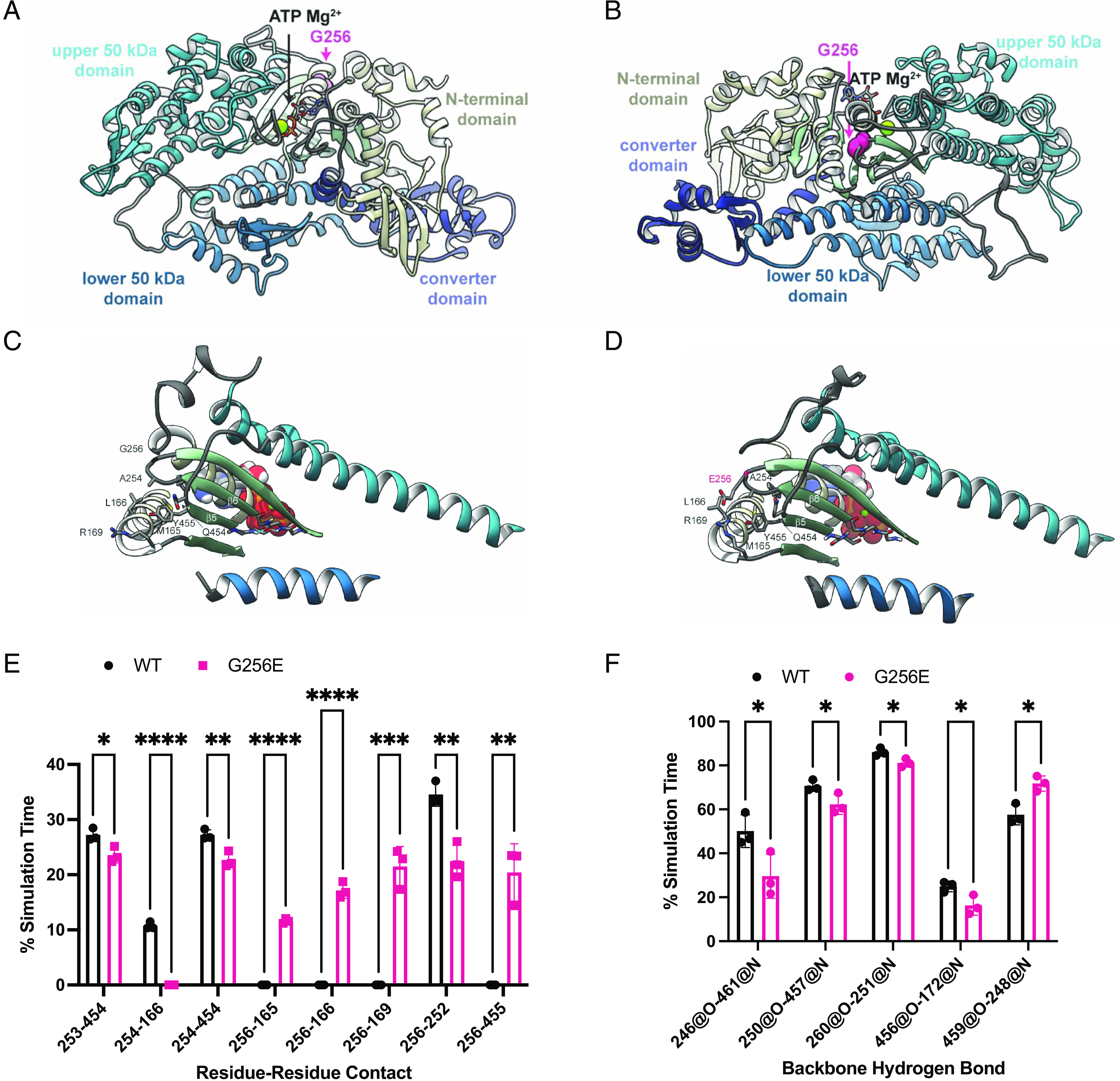
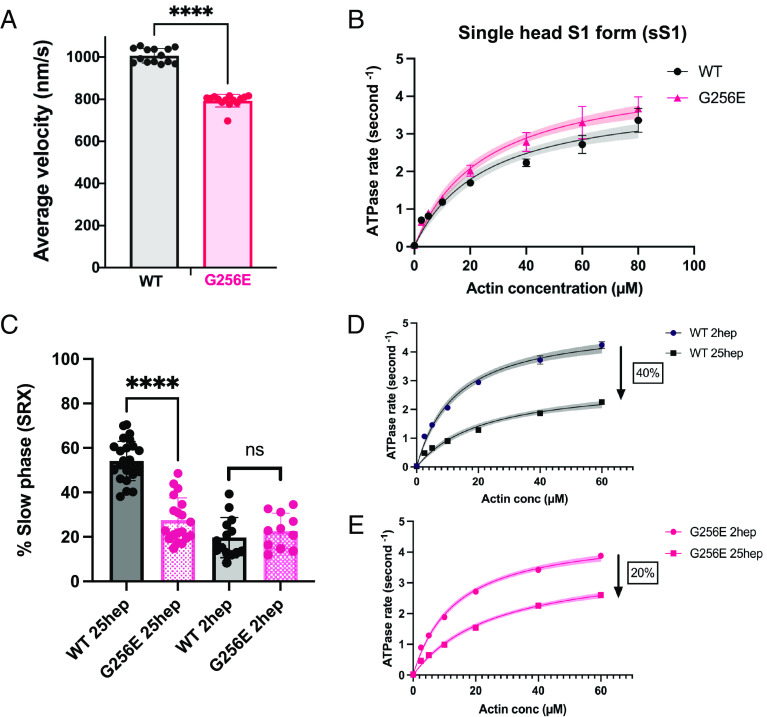
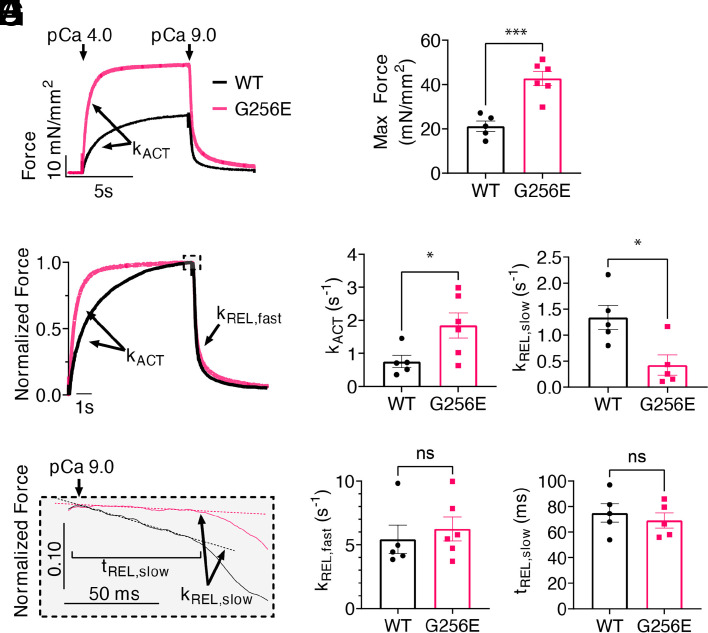
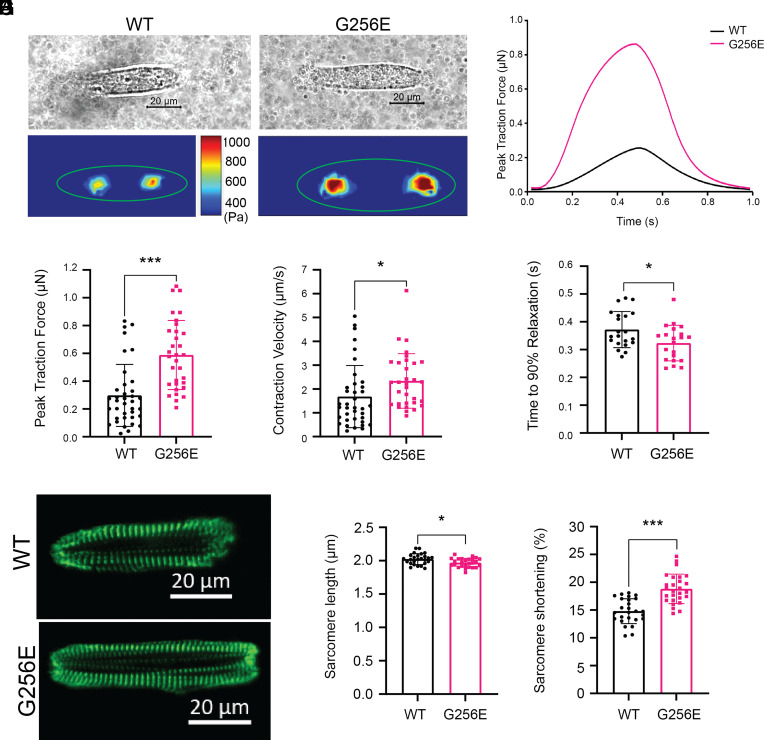
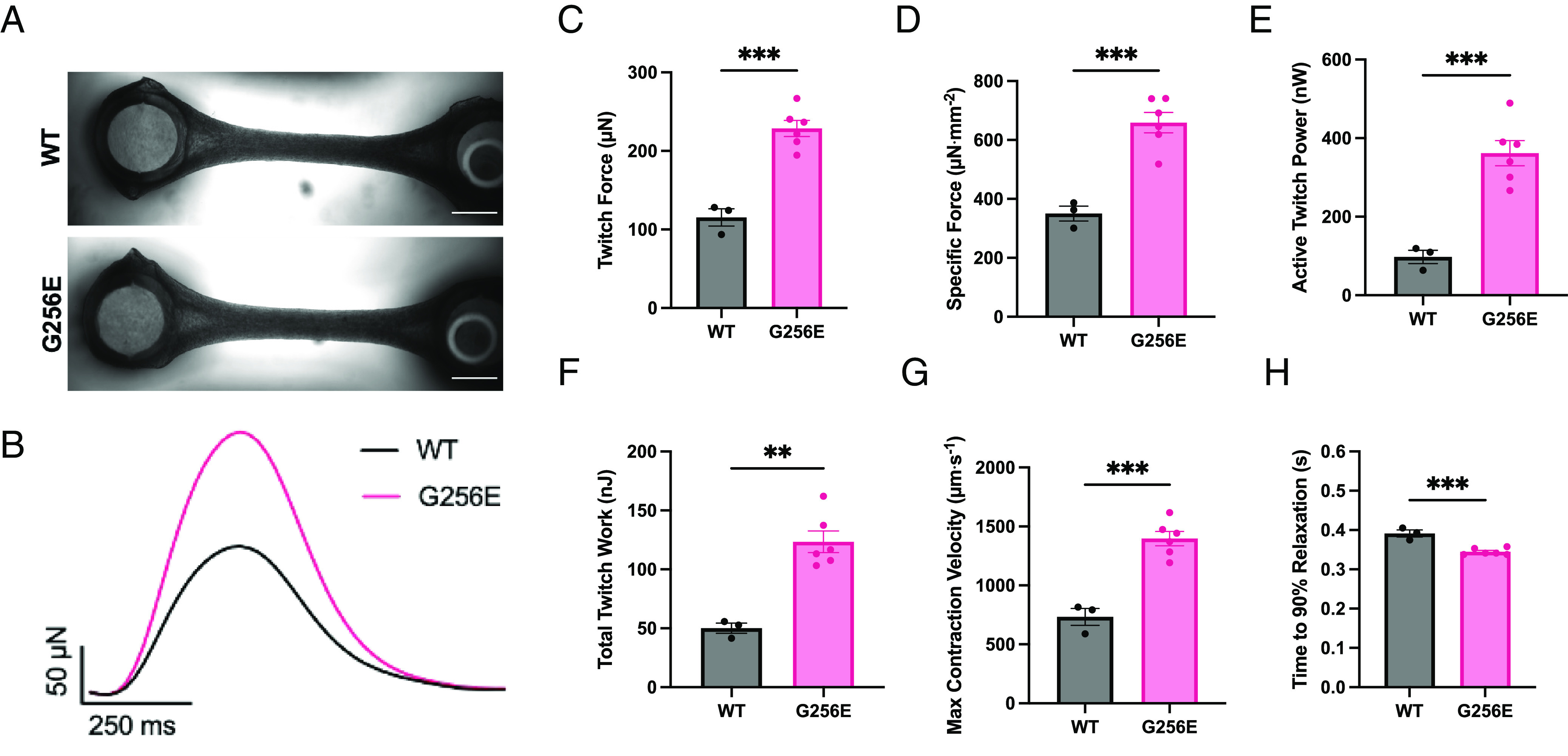
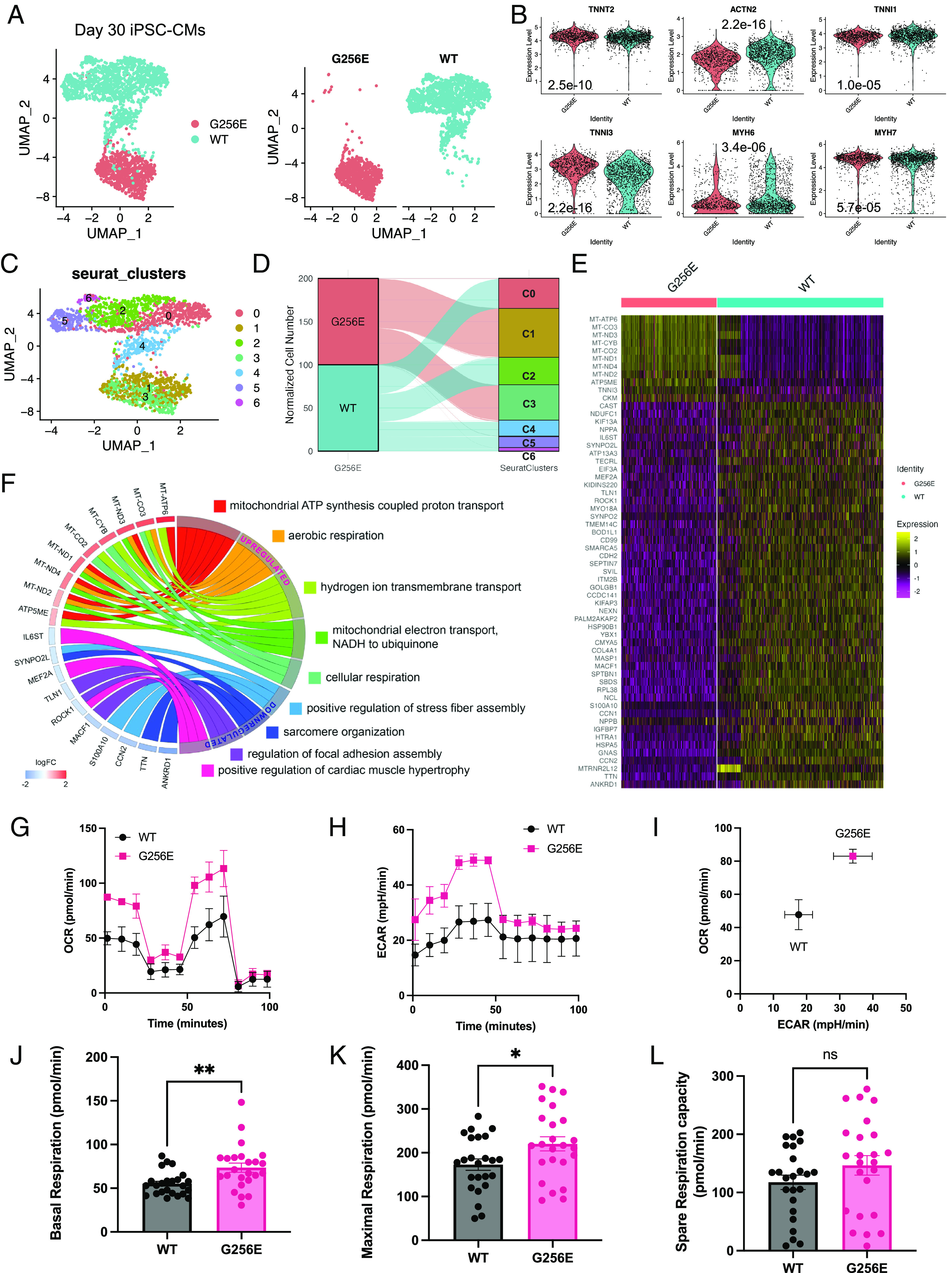
Determining the pathogenicity of hypertrophic cardiomyopathy-associated mutations in the β-myosin heavy chain (MYH7) can be challenging due to its variable penetrance and clinical severity. This study investigates the early pathogenic effects of the incomplete-penetrant MYH7 G256E mutation on myosin function that may trigger pathogenic adaptations and hypertrophy. We hypothesized that the G256E mutation would alter myosin biomechanical function, leading to changes in cellular functions. We developed a collaborative pipeline to characterize myosin function across protein, myofibril, cell, and tissue levels to determine the multiscale effects on structure-function of the contractile apparatus and its implications for gene regulation and metabolic state. The G256E mutation disrupts the transducer region of the S1 head and reduces the fraction of myosin in the folded-back state by 33%, resulting in more myosin heads available for contraction. Myofibrils from gene-edited MYH7WT/G256E human induced pluripotent stem cell-derived cardiomyocytes (hiPSC-CMs) exhibited greater and faster tension development. This hypercontractile phenotype persisted in single-cell hiPSC-CMs and engineered heart tissues. We demonstrated consistent hypercontractile myosin function as a primary consequence of the MYH7 G256E mutation across scales, highlighting the pathogenicity of this gene variant. Single-cell transcriptomic and metabolic profiling demonstrated upregulated mitochondrial genes and increased mitochondrial respiration, indicating early bioenergetic alterations. This work highlights the benefit of our multiscale platform to systematically evaluate the pathogenicity of gene variants at the protein and contractile organelle level and their early consequences on cellular and tissue function. We believe this platform can help elucidate the genotype-phenotype relationships underlying other genetic cardiovascular diseases.
Keywords: MYH7; biomechanics; hypertrophic cardiomyopathy; induced pluripotent stem cells.
Conflict of interest statement
Competing interests statement:J.A.S. is a co-founder and consultant for Cytokinetics Inc. and owns stock in the company, which has a focus on therapeutic treatments for cardiomyopathies and other muscle diseases. D.B. is a consultant for Cytokinetics Inc.
Figures
Update of
-
Multi-scale models reveal hypertrophic cardiomyopathy MYH7 G256E mutation drives hypercontractility and elevated mitochondrial respiration.bioRxiv [Preprint]. 2023 Jun 9:2023.06.08.544276. doi: 10.1101/2023.06.08.544276. bioRxiv. 2023. Update in: Proc Natl Acad Sci U S A. 2024 May 7;121(19):e2318413121. doi: 10.1073/pnas.2318413121. PMID: 37333118 Free PMC article. Updated. Preprint.
References
-
- Maron B. J., Maron M. S., Hypertrophic cardiomyopathy. Lancet 381, 242–255 (2013). - PubMed
-
- Semsarian C., Ingles J., Maron M. S., Maron B. J., New perspectives on the prevalence of hypertrophic cardiomyopathy. J. Am. Coll. Cardiol. 65, 1249–1254 (2015). - PubMed
-
- Alfares A. A., et al. , Results of clinical genetic testing of 2,912 probands with hypertrophic cardiomyopathy: Expanded panels offer limited additional sensitivity. Genet. Med. 17, 880–888 (2015). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
