

A secondary mechanism of action for triazole antifungals in Aspergillus fumigatus mediated by hmg1
- PMID: 38684680
- PMCID: PMC11059170
- DOI: 10.1038/s41467-024-48029-2
A secondary mechanism of action for triazole antifungals in Aspergillus fumigatus mediated by hmg1
Abstract
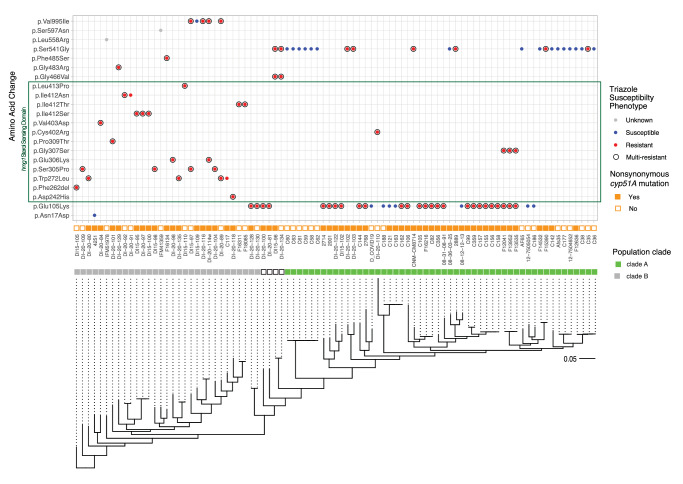
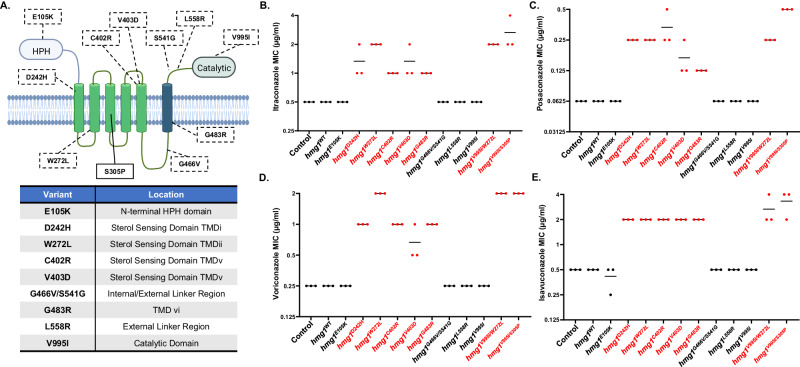
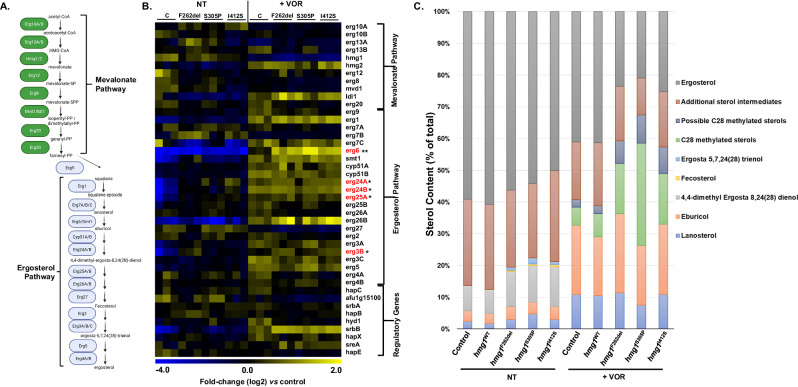
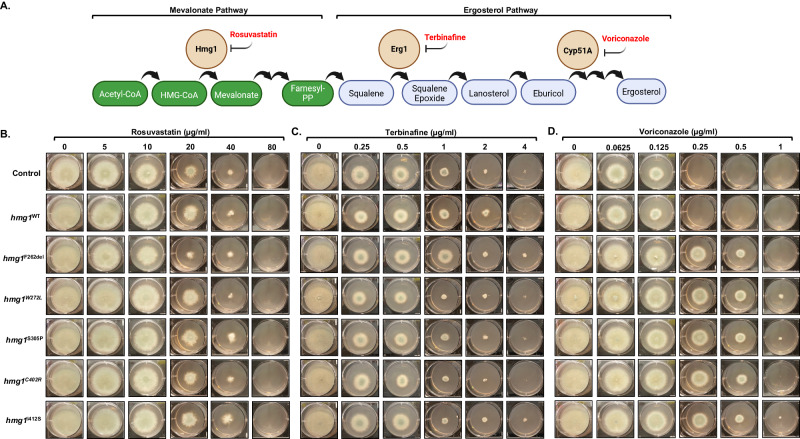
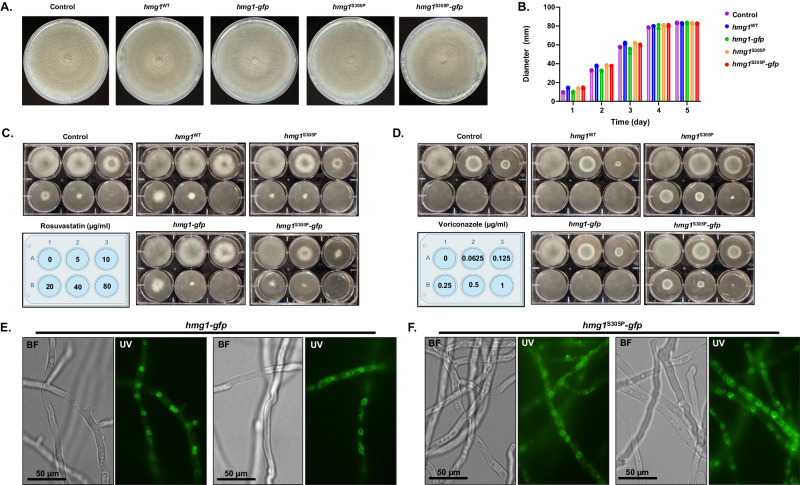
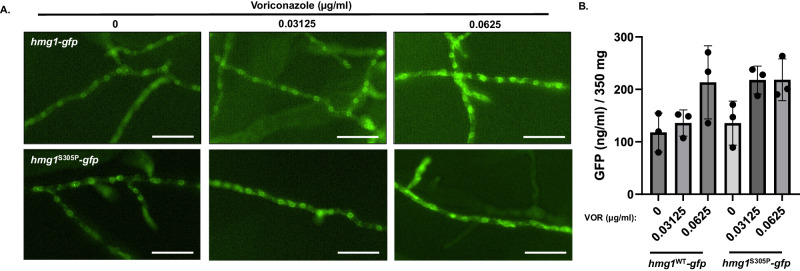
Triazole antifungals function as ergosterol biosynthesis inhibitors and are frontline therapy for invasive fungal infections, such as invasive aspergillosis. The primary mechanism of action of triazoles is through the specific inhibition of a cytochrome P450 14-α-sterol demethylase enzyme, Cyp51A/B, resulting in depletion of cellular ergosterol. Here, we uncover a clinically relevant secondary mechanism of action for triazoles within the ergosterol biosynthesis pathway. We provide evidence that triazole-mediated inhibition of Cyp51A/B activity generates sterol intermediate perturbations that are likely decoded by the sterol sensing functions of HMG-CoA reductase and Insulin-Induced Gene orthologs as increased pathway activity. This, in turn, results in negative feedback regulation of HMG-CoA reductase, the rate-limiting step of sterol biosynthesis. We also provide evidence that HMG-CoA reductase sterol sensing domain mutations previously identified as generating resistance in clinical isolates of Aspergillus fumigatus partially disrupt this triazole-induced feedback. Therefore, our data point to a secondary mechanism of action for the triazoles: induction of HMG-CoA reductase negative feedback for downregulation of ergosterol biosynthesis pathway activity. Abrogation of this feedback through acquired mutations in the HMG-CoA reductase sterol sensing domain diminishes triazole antifungal activity against fungal pathogens and underpins HMG-CoA reductase-mediated resistance.
© 2024. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
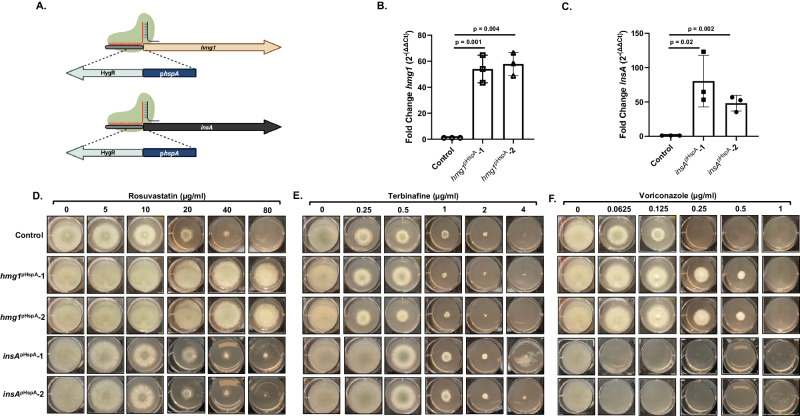
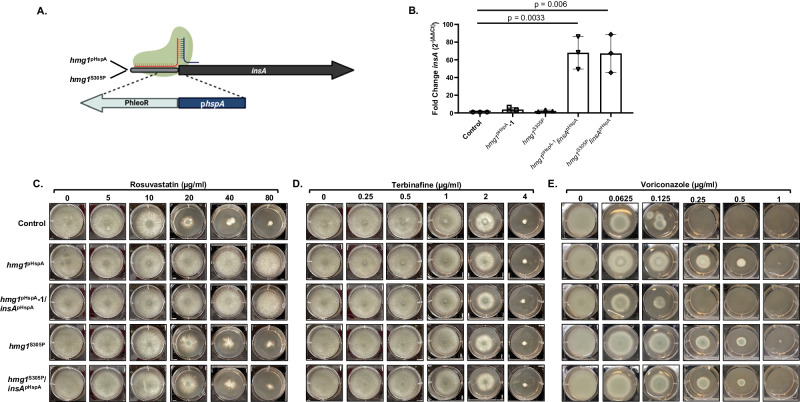
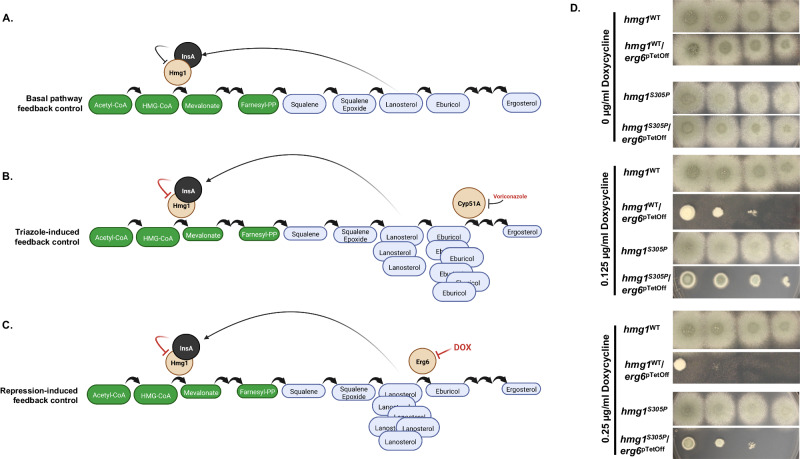
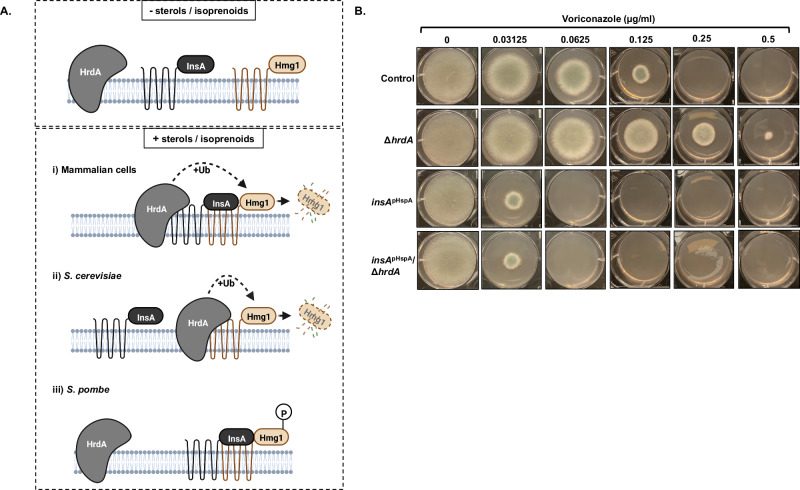
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
