

Identification and affinity enhancement of T-cell receptor targeting a KRASG12V cancer neoantigen
- PMID: 38684865
- PMCID: PMC11058820
- DOI: 10.1038/s42003-024-06209-2
Identification and affinity enhancement of T-cell receptor targeting a KRASG12V cancer neoantigen
Abstract
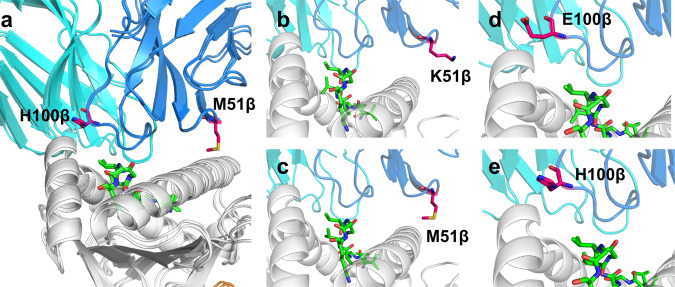
Neoantigens derived from somatic mutations in Kirsten Rat Sarcoma Viral Oncogene Homolog (KRAS), the most frequently mutated oncogene, represent promising targets for cancer immunotherapy. Recent research highlights the potential role of human leukocyte antigen (HLA) allele A*11:01 in presenting these altered KRAS variants to the immune system. In this study, we successfully generate and identify murine T-cell receptors (TCRs) that specifically recognize KRAS8-16G12V from three predicted high affinity peptides. By determining the structure of the tumor-specific 4TCR2 bound to KRASG12V-HLA-A*11:01, we conduct structure-based design to create and evaluate TCR variants with markedly enhanced affinity, up to 15.8-fold. This high-affinity TCR mutant, which involved only two amino acid substitutions, display minimal conformational alterations while maintaining a high degree of specificity for the KRASG12V peptide. Our research unveils the molecular mechanisms governing TCR recognition towards KRASG12V neoantigen and yields a range of affinity-enhanced TCR mutants with significant potential for immunotherapy strategies targeting tumors harboring the KRASG12V mutation.
© 2024. The Author(s).
Conflict of interest statement
M.Z., F.G., F.W., and S.Y. are listed as inventors for a patent granted to generate TCR variant towards KRAS8–16G12V–HLA-A*11:01. The remaining authors declare no competing interests.
Figures
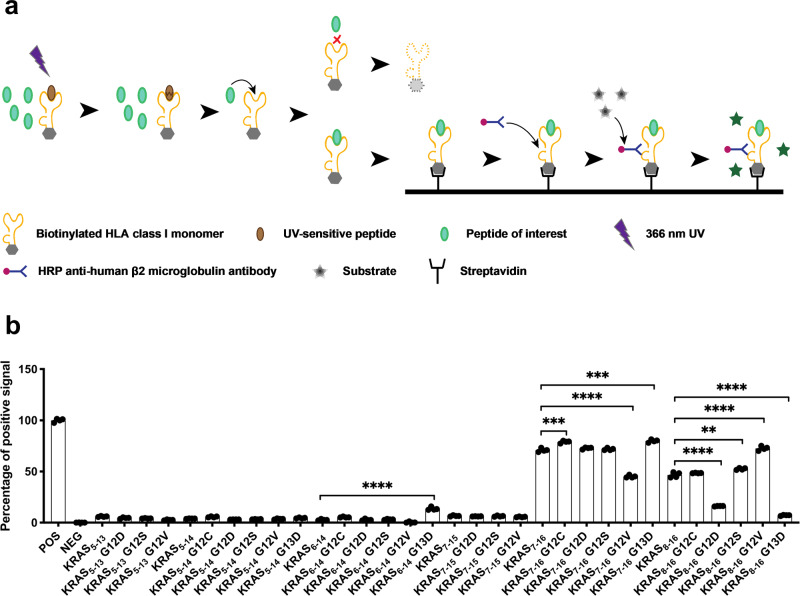
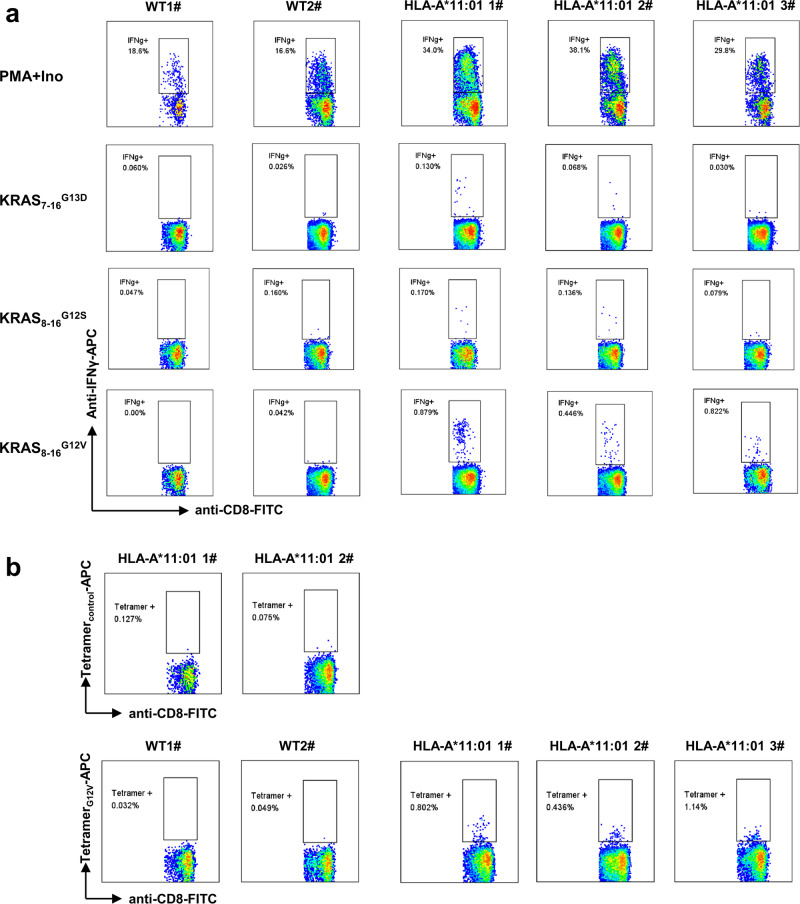
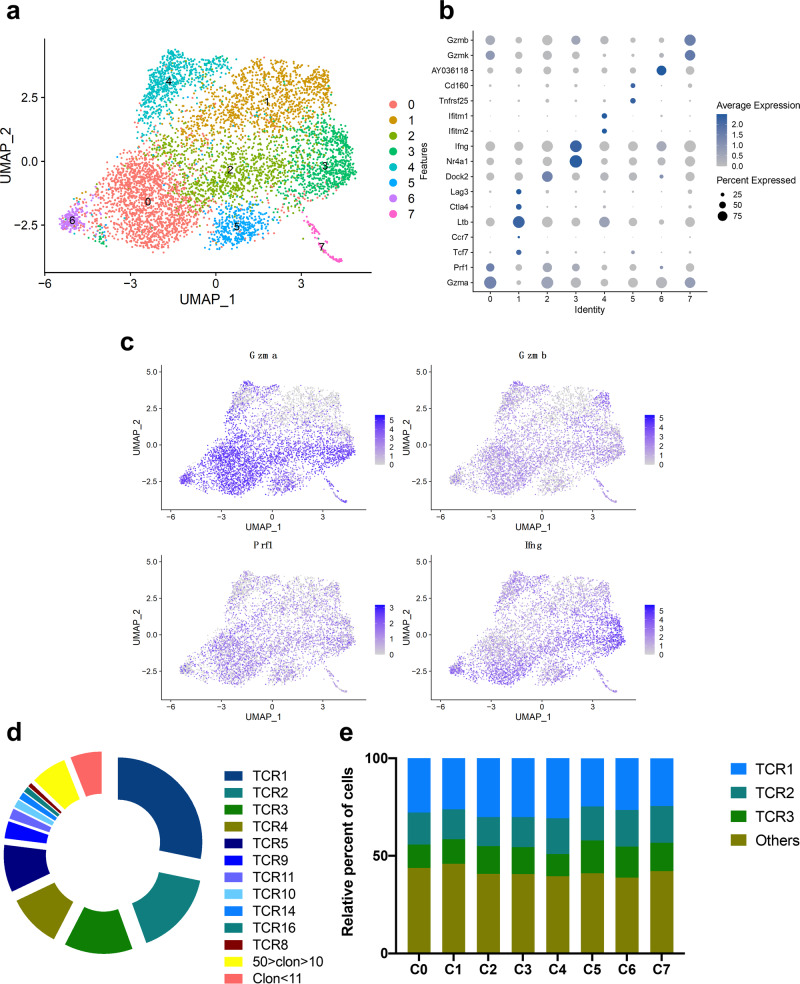
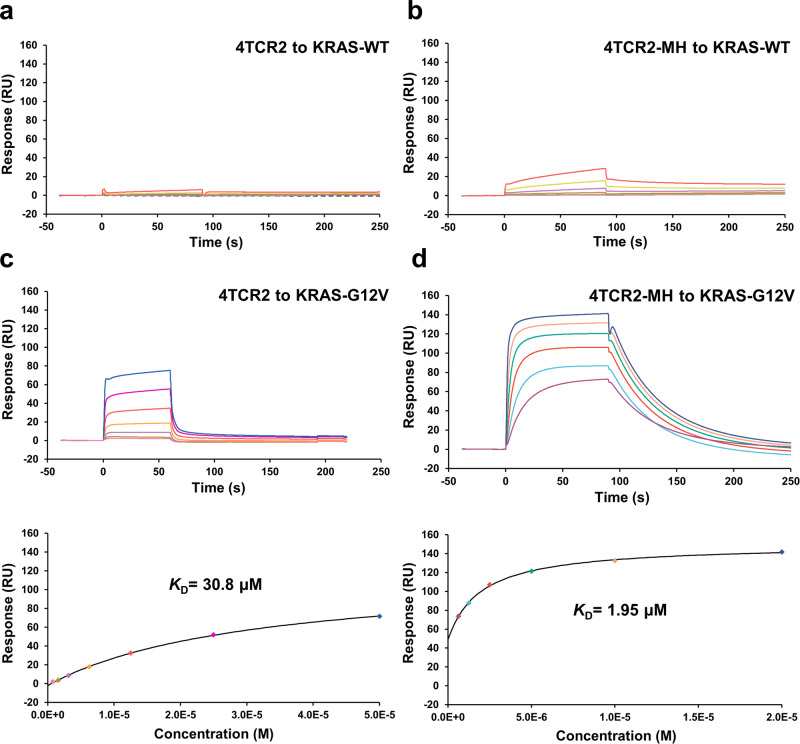


Similar articles
-
Therapeutic potential of T-cell receptor targeting the HLA-A*11:01-restricted KRASG12V neoantigen without cross-recognition of the self-antigen RAB7B in solid tumors.J Immunother Cancer. 2025 Jul 18;13(7):e011863. doi: 10.1136/jitc-2025-011863. J Immunother Cancer. 2025. PMID: 40681176 Free PMC article.
-
Identification and structural characterization of a mutant KRAS-G12V specific TCR restricted by HLA-A3.Eur J Immunol. 2024 Sep;54(9):e2451079. doi: 10.1002/eji.202451079. Epub 2024 Jul 18. Eur J Immunol. 2024. PMID: 39030753
-
Targeting KRASG12V mutations with HLA class II-restricted TCR for the immunotherapy in solid tumors.Front Immunol. 2023 May 23;14:1161538. doi: 10.3389/fimmu.2023.1161538. eCollection 2023. Front Immunol. 2023. PMID: 37287989 Free PMC article.
-
Facts and Hopes in Immunotherapy Strategies Targeting Antigens Derived from KRAS Mutations.Clin Cancer Res. 2024 May 15;30(10):2017-2024. doi: 10.1158/1078-0432.CCR-23-1212. Clin Cancer Res. 2024. PMID: 38266167 Free PMC article. Review.
-
Structural basis for T cell recognition of cancer neoantigens and implications for predicting neoepitope immunogenicity.Front Immunol. 2023 Nov 17;14:1303304. doi: 10.3389/fimmu.2023.1303304. eCollection 2023. Front Immunol. 2023. PMID: 38045695 Free PMC article. Review.
Cited by
-
Focusing on CD8+ T-cell phenotypes: improving solid tumor therapy.J Exp Clin Cancer Res. 2024 Sep 28;43(1):266. doi: 10.1186/s13046-024-03195-5. J Exp Clin Cancer Res. 2024. PMID: 39342365 Free PMC article. Review.
-
Cell-based immunotherapies for solid tumors: advances, challenges, and future directions.Front Oncol. 2025 Apr 28;15:1551583. doi: 10.3389/fonc.2025.1551583. eCollection 2025. Front Oncol. 2025. PMID: 40356763 Free PMC article. Review.
-
The Evolving T Cell Receptor Recognition Code: The Rules Are More Like Guidelines.Immunol Rev. 2025 Jan;329(1):e13439. doi: 10.1111/imr.13439. Immunol Rev. 2025. PMID: 39804137 Review.
-
Safety and Tolerability of Letetresgene Autoleucel (GSK3377794): Pilot Studies in Patients with Advanced Non-Small Cell Lung Cancer.Clin Cancer Res. 2025 Feb 3;31(3):529-542. doi: 10.1158/1078-0432.CCR-24-1591. Clin Cancer Res. 2025. PMID: 39576208 Free PMC article. Clinical Trial.
-
Shared neoantigens for cancer immunotherapy.Mol Ther Oncol. 2025 Mar 28;33(2):200978. doi: 10.1016/j.omton.2025.200978. eCollection 2025 Jun 18. Mol Ther Oncol. 2025. PMID: 40256120 Free PMC article. Review.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
