

IL-23 past, present, and future: a roadmap to advancing IL-23 science and therapy
- PMID: 38686385
- PMCID: PMC11056518
- DOI: 10.3389/fimmu.2024.1331217
IL-23 past, present, and future: a roadmap to advancing IL-23 science and therapy
Abstract
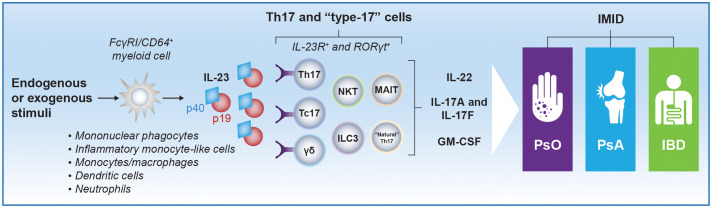
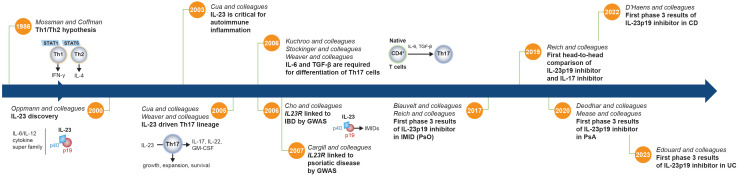
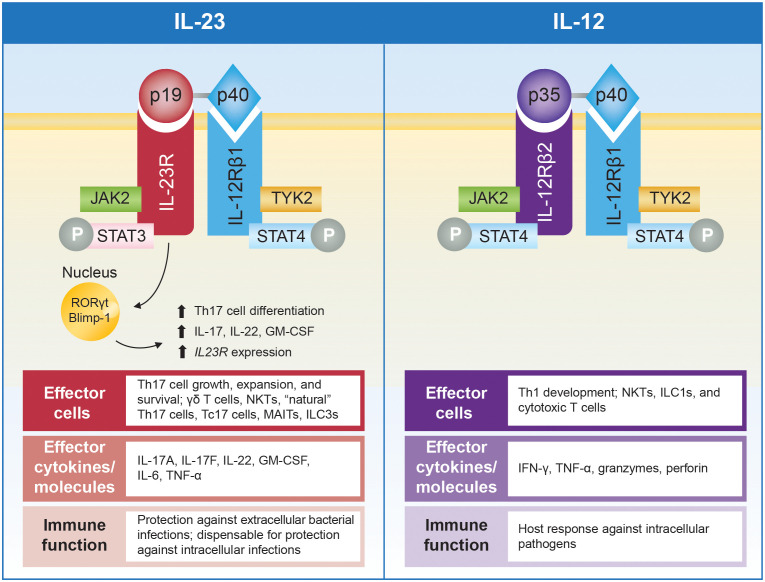
Interleukin (IL)-23, an IL-12 cytokine family member, is a hierarchically dominant regulatory cytokine in a cluster of immune-mediated inflammatory diseases (IMIDs), including psoriasis, psoriatic arthritis, and inflammatory bowel disease. We review IL-23 biology, IL-23 signaling in IMIDs, and the effect of IL-23 inhibition in treating these diseases. We propose studies to advance IL-23 biology and unravel differences in response to anti-IL-23 therapy. Experimental evidence generated from these investigations could establish a novel molecular ontology centered around IL-23-driven diseases, improve upon current approaches to treating IMIDs with IL-23 inhibition, and ultimately facilitate optimal identification of patients and, thereby, outcomes.
Keywords: IL-23; cytokine; immune-mediated inflammatory diseases; inflammatory bowel disease; psoriasis; psoriatic arthritis.
Copyright © 2024 Krueger, Eyerich, Kuchroo, Ritchlin, Abreu, Elloso, Fourie, Fakharzadeh, Sherlock, Yang, Cua and McInnes.
Conflict of interest statement
JK served as a consultant for and/or received honoraria from AbbVie, Allergan, Almirall, Amgen, Arena, Aristea, Asana, Aurigene, Biogen Idec, Boehringer Ingelheim, Bristol Myers Squibb, Escalier, Galapagos, Janssen, Eli Lilly, MoonLake Immunotherapeutics, Nimbus, Novartis, Pfizer, Sanofi, Sienna Biopharmaceuticals, Sun Pharmaceutical Industries, Target-Derm, UCB, Valeant, and Ventyx. KE received speaker fees from and/or served on an advisory board for AbbVie, Almirall, Boehringer Ingelheim, Bristol Myers Squibb, Galapagos, Eli Lilly, Janssen, Pfizer, Novartis, Sanofi, and UCB. VK served as a consultant for iTeos; has an ownership interest in and is a member of the scientific advisory board for Tizona Therapeutics; is a cofounder of and has an ownership interest in Celsius Therapeutics; and is a cofounder of Bicara Therapeutics. CR received grant/research support and consulting fees from UCB, AbbVie, and Amgen; and received consulting fees from Eli Lilly, Pfizer, Novartis, Gilead, and Janssen. MA is a consultant or served on advisory boards for AbbVie Inc, Arena Pharmaceuticals Inc now Pfizer, Bristol Myers Squibb, Celsius Therapeutics, Eli Lilly and Company, Gilead Sciences Inc, Janssen Pharmaceuticals, Janssen Global Services, Pfizer Pharmaceutical, Prometheus Biosciences, UCB Biopharma SRL. She has received fees for lecturing from Alimentiv, Janssen Pharmaceuticals, Prime CME and WebMD Global LLC. IM received consulting fees and grant/research support from AstraZeneca, Bristol Myers Squibb, Amgen, Eli Lilly, GSK, Janssen, Novartis, Roche, and UCB; received consulting fees from AbbVie, Cabaletta, Compugen, Gilead, Pfizer, and Sanofi; serves as a shareholder of Compugen and Causeway Therapeutics, and as a board member for National Health Service Greater Glasgow and Clyde; and is a trustee for Versus Arthritis. ME, AF, SF, JS, Y-WY, and DC are employees of Janssen and hold stock in Johnson & Johnson. Box 1Questions to consider for future investigation to advance the science of IL-23 and approaches to IL-23 inhibitor therapy.A multiomics approach to advancing the science of IL-23 Are there any trends in the literature that identify how the IL-23/IL-17 axis may function and drive nuanced effects in different cell types or tissues?Does the impact of this pathway change over time or with progression of disease, identifying an ideal timeframe or context in the course of disease to initiate IL-23 inhibitor treatment?Are there any differences in biomarker profiles between inadequate responders and responders to therapy that may be used to predict clinical outcomes?What is the epigenetic profile of IL-23 signaling in various immune and non-immune cell types and how may this impact disease pathogenesis, disease progression, or response to treatment?What post-translational modifications of downstream cytokines/effector molecules are associated with IL-23 signaling?What are the functional consequences of changes in multiomics profiles and associated changes in immune cell populations over the course of disease and in response to treatment?“Broad sweep” of IL-23 receptor–expressing cells Which cells express the IL-23 receptor in healthy and inflamed tissues in humans?What is the role of IL-23 receptor signaling in diverse T-cell targets?How may a comprehensive assessment of IL-23 receptor expression across cell types and tissues identify new disease states that may be treatable through targeted intervention with IL-23 inhibition?How does IL-23 receptor expression change at various timepoints and tissue types across IMIDs and in response to treatment with IL-23 blockade?Is the IL-23 receptor coexpressed with other molecules of interest (ie, other cytokines or receptors) that may help to explain differences between currently available therapies and help design future treatments?Gaps in understanding IL-23 signaling and cellular activity Are there different nuances to IL-23 signaling in different cell types or tissues that may have functional consequences impacting disease pathogenesis or response to treatment?Do these signaling nuances change over the course of disease progression, and normalize with treatment with an IL-23 inhibitor?What is the role of STAT3 and STAT4 signaling in the context of IL-23 receptor signaling in Treg cells?What is the relationship between tissue-specific microbiota and IL-23 receptor signaling?Durability of response What is the effect of targeting IL-23p19 on TRM cells and the ratio of TRM/Treg cells across IMIDs?What are the mechanisms underlying the long-lasting effects of targeting IL-23p19?Can early intervention with IL-23 blockade after recent onset of disease modify the course of disease progression by suppressing TRM cells?Future IL-23 inhibitor molecules What molecular attributes of IL-23 inhibitors may be relevant for optimizing inhibition of IL-23 across IMIDs?Are there therapeutic advantages in targeting the IL-23 receptor versus IL-23p19 subunit directly in treating IMIDs?Combination therapy What potential therapeutic benefits may derive from combining IL-23 inhibition with blockade of another complementary inflammatory disease-driving pathway in treating IMIDs?What complementary pathway(s) would be optimal for blockade in combination with IL-23 inhibition for treatment of given IMIDs?IL, interleukin; STAT, signal transducer and activator of transcription; Treg, regulatory T.
Figures
References
-
- Blauvelt A, Papp KA, Griffiths CE, Randazzo B, Wasfi Y, Shen YK, et al. Efficacy and safety of guselkumab, an anti-interleukin-23 monoclonal antibody, compared with adalimumab for the continuous treatment of patients with moderate to severe psoriasis: results from the phase III, double-blinded, placebo- and active comparator-controlled VOYAGE 1 trial. J Am Acad Dermatol. (2017) 76:405–17. doi: 10.1016/j.jaad.2016.11.041 - DOI - PubMed
-
- Reich K, Armstrong AW, Foley P, Song M, Wasfi Y, Randazzo B, et al. Efficacy and safety of guselkumab, an anti-interleukin-23 monoclonal antibody, compared with adalimumab for the treatment of patients with moderate to severe psoriasis with randomized withdrawal and retreatment: results from the phase III, double-blind, placebo- and active comparator-controlled VOYAGE 2 trial. J Am Acad Dermatol. (2017) 76:418–31. doi: 10.1016/j.jaad.2016.11.042 - DOI - PubMed
-
- Deodhar A, Helliwell PS, Boehncke WH, Kollmeier AP, Hsia EC, Subramanian RA, et al. Guselkumab in patients with active psoriatic arthritis who were biologic-naive or had previously received TNFα inhibitor treatment (DISCOVER-1): a double-blind, randomised, placebo-controlled phase 3 trial. Lancet. (2020) 395:1115–25. doi: 10.1016/s0140-6736(20)30265-8 - DOI - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous