

All Three Supersystems-Nervous, Vascular, and Immune-Contribute to the Cortical Infarcts After Subarachnoid Hemorrhage
- PMID: 38689162
- PMCID: PMC11772491
- DOI: 10.1007/s12975-024-01242-z
All Three Supersystems-Nervous, Vascular, and Immune-Contribute to the Cortical Infarcts After Subarachnoid Hemorrhage
Abstract
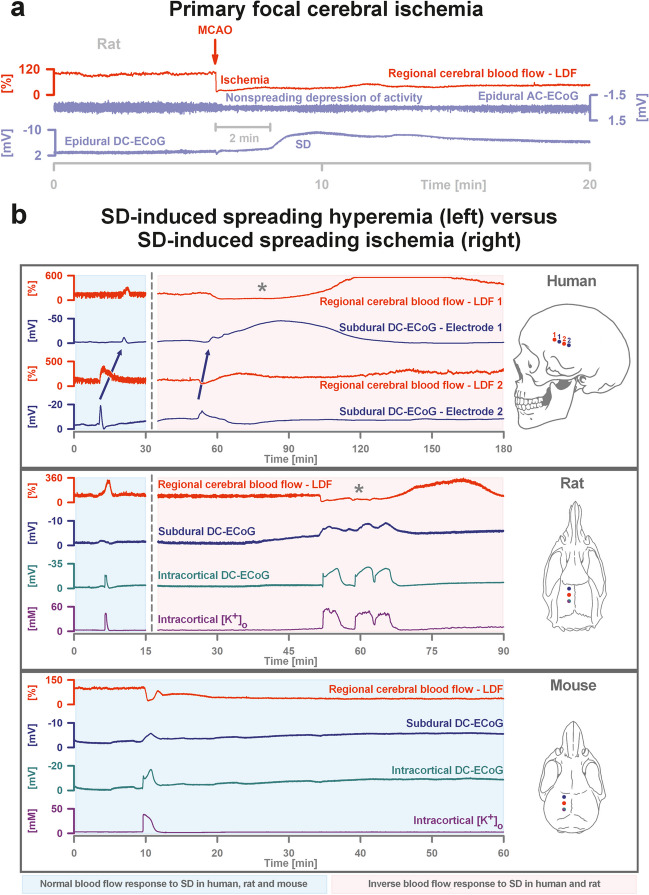
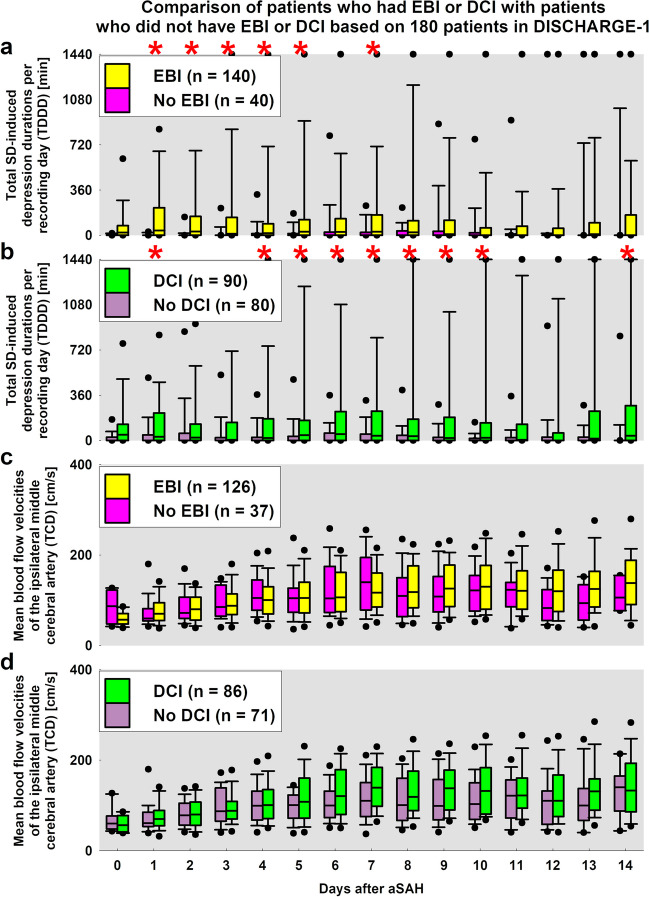
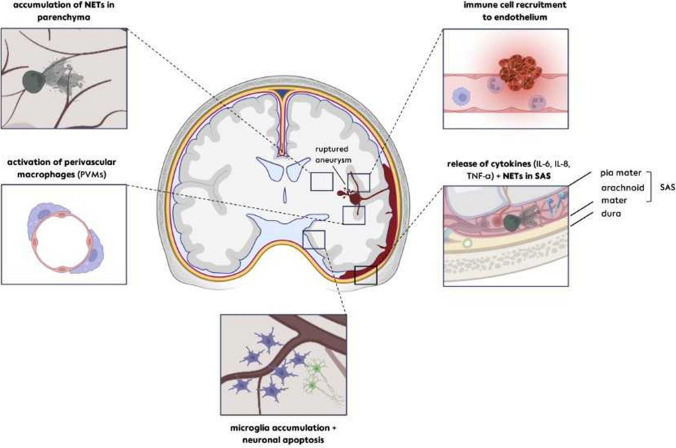
The recently published DISCHARGE-1 trial supports the observations of earlier autopsy and neuroimaging studies that almost 70% of all focal brain damage after aneurysmal subarachnoid hemorrhage are anemic infarcts of the cortex, often also affecting the white matter immediately below. The infarcts are not limited by the usual vascular territories. About two-fifths of the ischemic damage occurs within ~ 48 h; the remaining three-fifths are delayed (within ~ 3 weeks). Using neuromonitoring technology in combination with longitudinal neuroimaging, the entire sequence of both early and delayed cortical infarct development after subarachnoid hemorrhage has recently been recorded in patients. Characteristically, cortical infarcts are caused by acute severe vasospastic events, so-called spreading ischemia, triggered by spontaneously occurring spreading depolarization. In locations where a spreading depolarization passes through, cerebral blood flow can drastically drop within a few seconds and remain suppressed for minutes or even hours, often followed by high-amplitude, sustained hyperemia. In spreading depolarization, neurons lead the event, and the other cells of the neurovascular unit (endothelium, vascular smooth muscle, pericytes, astrocytes, microglia, oligodendrocytes) follow. However, dysregulation in cells of all three supersystems-nervous, vascular, and immune-is very likely involved in the dysfunction of the neurovascular unit underlying spreading ischemia. It is assumed that subarachnoid blood, which lies directly on the cortex and enters the parenchyma via glymphatic channels, triggers these dysregulations. This review discusses the neuroglial, neurovascular, and neuroimmunological dysregulations in the context of spreading depolarization and spreading ischemia as critical elements in the pathogenesis of cortical infarcts after subarachnoid hemorrhage.
Keywords: Delayed cerebral ischemia; Neuromonitoring; Spreading depolarization; Stroke; Subarachnoid hemorrhage; Vasospasm.
© 2024. The Author(s).
Conflict of interest statement
Declarations. Competing interests: The authors declare no competing interests. Ethical Approval: All procedures performed involving patients were in accordance with the ethical standards of the institutional research committees and with the 1964 Helsinki Declaration and its later amendments. Either informed consent or surrogate informed consent was obtained for all patients. Conflict of Interest: The authors declare no competing interests.
Figures
Similar articles
-
Similarities in the Electrographic Patterns of Delayed Cerebral Infarction and Brain Death After Aneurysmal and Traumatic Subarachnoid Hemorrhage.Transl Stroke Res. 2025 Feb;16(1):147-168. doi: 10.1007/s12975-024-01237-w. Epub 2024 Feb 23. Transl Stroke Res. 2025. PMID: 38396252 Free PMC article. Review.
-
Requisite ischemia for spreading depolarization occurrence after subarachnoid hemorrhage in rodents.J Cereb Blood Flow Metab. 2017 May;37(5):1829-1840. doi: 10.1177/0271678X16659303. Epub 2016 Jan 1. J Cereb Blood Flow Metab. 2017. PMID: 27432225 Free PMC article.
-
Subarachnoid blood acutely induces spreading depolarizations and early cortical infarction.Brain. 2017 Oct 1;140(10):2673-2690. doi: 10.1093/brain/awx214. Brain. 2017. PMID: 28969382 Free PMC article.
-
Report of selective cortical infarcts in the primate clot model of vasospasm after subarachnoid hemorrhage.Neurosurgery. 2010 Sep;67(3):721-8; discussion 728-9. doi: 10.1227/01.NEU.0000378024.70848.8F. Neurosurgery. 2010. PMID: 20651629
-
Cerebrovascular pathophysiology of delayed cerebral ischemia after aneurysmal subarachnoid hemorrhage.Histol Histopathol. 2021 Feb;36(2):143-158. doi: 10.14670/HH-18-253. Epub 2020 Sep 30. Histol Histopathol. 2021. PMID: 32996580 Review.
Cited by
-
Targeted proteomic analysis identifies ACVRL1 as a marker of cerebral edema in aneurysmal subarachnoid hemorrhage.J Cereb Blood Flow Metab. 2025 Aug 20:271678X251361250. doi: 10.1177/0271678X251361250. Online ahead of print. J Cereb Blood Flow Metab. 2025. PMID: 40832973 Free PMC article.
-
MiR-34c Is Predictive of Delayed Cerebral Ischemia After Subarachnoid Hemorrhage.Res Sq [Preprint]. 2025 Apr 1:rs.3.rs-6198784. doi: 10.21203/rs.3.rs-6198784/v1. Res Sq. 2025. PMID: 40235490 Free PMC article. Preprint.
-
Subarachnoid hemorrhage distinctively disrupts the glymphatic and meningeal lymphatic systems in beagles.Theranostics. 2024 Sep 16;14(15):6053-6070. doi: 10.7150/thno.100982. eCollection 2024. Theranostics. 2024. PMID: 39346537 Free PMC article.
-
Oxygen-Based Autoregulation Indices Associated with Clinical Outcomes and Spreading Depolarization in Aneurysmal Subarachnoid Hemorrhage.Neurocrit Care. 2025 Apr;42(2):521-531. doi: 10.1007/s12028-024-02088-x. Epub 2024 Aug 27. Neurocrit Care. 2025. PMID: 39192101 Free PMC article.
-
Angiographic perfusion outperforms large artery vasospasm for predicting the impact of rescue therapy in subarachnoid hemorrhage.J Cereb Blood Flow Metab. 2025 Jul 28:271678X251361992. doi: 10.1177/0271678X251361992. Online ahead of print. J Cereb Blood Flow Metab. 2025. PMID: 40719609 Free PMC article.
References
-
- Connolly ES Jr, Rabinstein AA, Carhuapoma JR, Derdeyn CP, Dion J, Higashida RT, et al. Guidelines for the management of aneurysmal subarachnoid hemorrhage: a guideline for healthcare professionals from the American Heart Association/American Stroke Association. Stroke. 2012;43(6):1711–37. 10.1161/STR.0b013e3182587839. - PubMed
-
- Dreier JP, Winkler MKL, Major S, Horst V, Lublinsky S, Kola V, et al. Spreading depolarizations in ischaemia after subarachnoid haemorrhage, a diagnostic phase III study. Brain. 2022;145(4):1264–84. 10.1093/brain/awab457. - PubMed
-
- Birse SH, Tom MI. Incidence of cerebral infarction associated with ruptured intracranial aneurysms. A study of 8 unoperated cases of anterior cerebral aneurysm. Neurology. 1960;10:101–6. - PubMed
-
- Dreier JP, Sakowitz OW, Harder A, Zimmer C, Dirnagl U, Valdueza JM, Unterberg AW. Focal laminar cortical MR signal abnormalities after subarachnoid hemorrhage. Ann Neurol. 2002;52(6):825–9. 10.1002/ana.10383. - PubMed
Publication types
MeSH terms
Grants and funding
- R01 NS115887/NS/NINDS NIH HHS/United States
- Innovation Cluster Grant/German Center for Cardiovascular Research
- BIH-Charité Clinician Scientist Program/Charité - Universitätsmedizin Berlin and the Berlin Institute of Health
- DFG DR 323/10-2/Deutsche Forschungsgemeinschaft
- 1R01NS115887/NH/NIH HHS/United States
LinkOut - more resources
Full Text Sources