

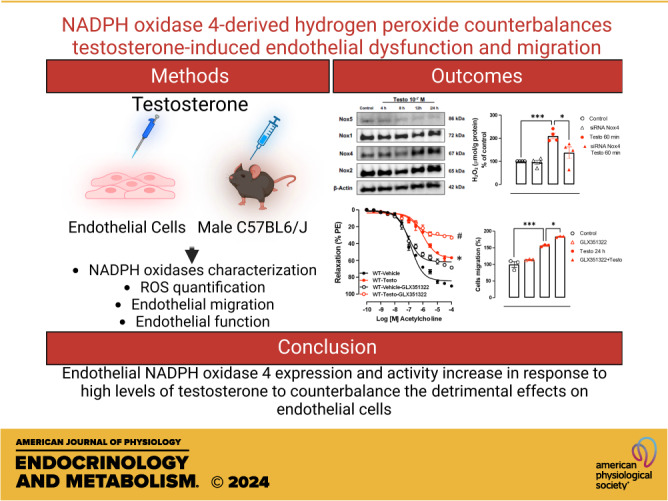
NADPH oxidase 4-derived hydrogen peroxide counterbalances testosterone-induced endothelial dysfunction and migration
- PMID: 38690939
- PMCID: PMC11390122
- DOI: 10.1152/ajpendo.00365.2023
NADPH oxidase 4-derived hydrogen peroxide counterbalances testosterone-induced endothelial dysfunction and migration
Abstract
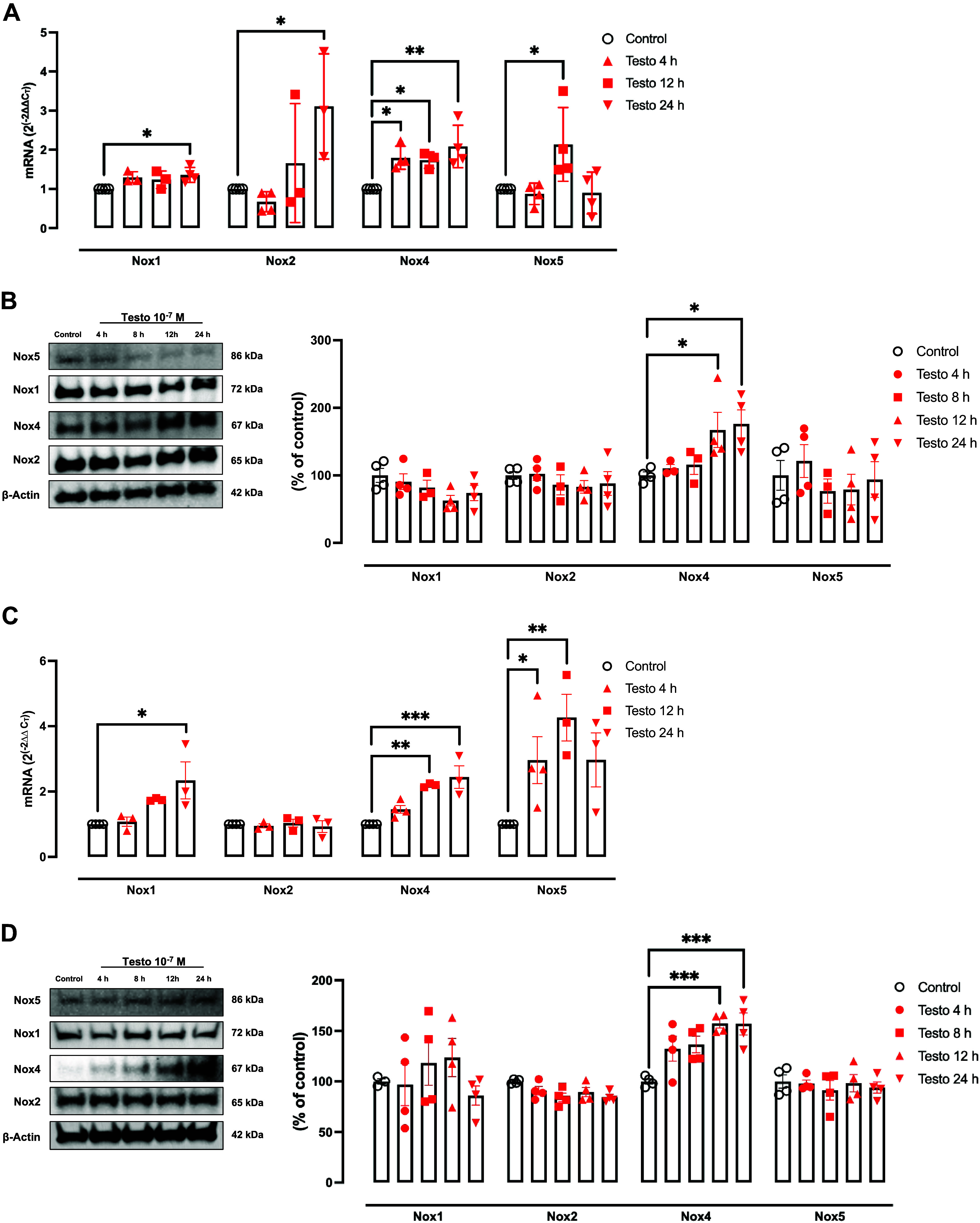
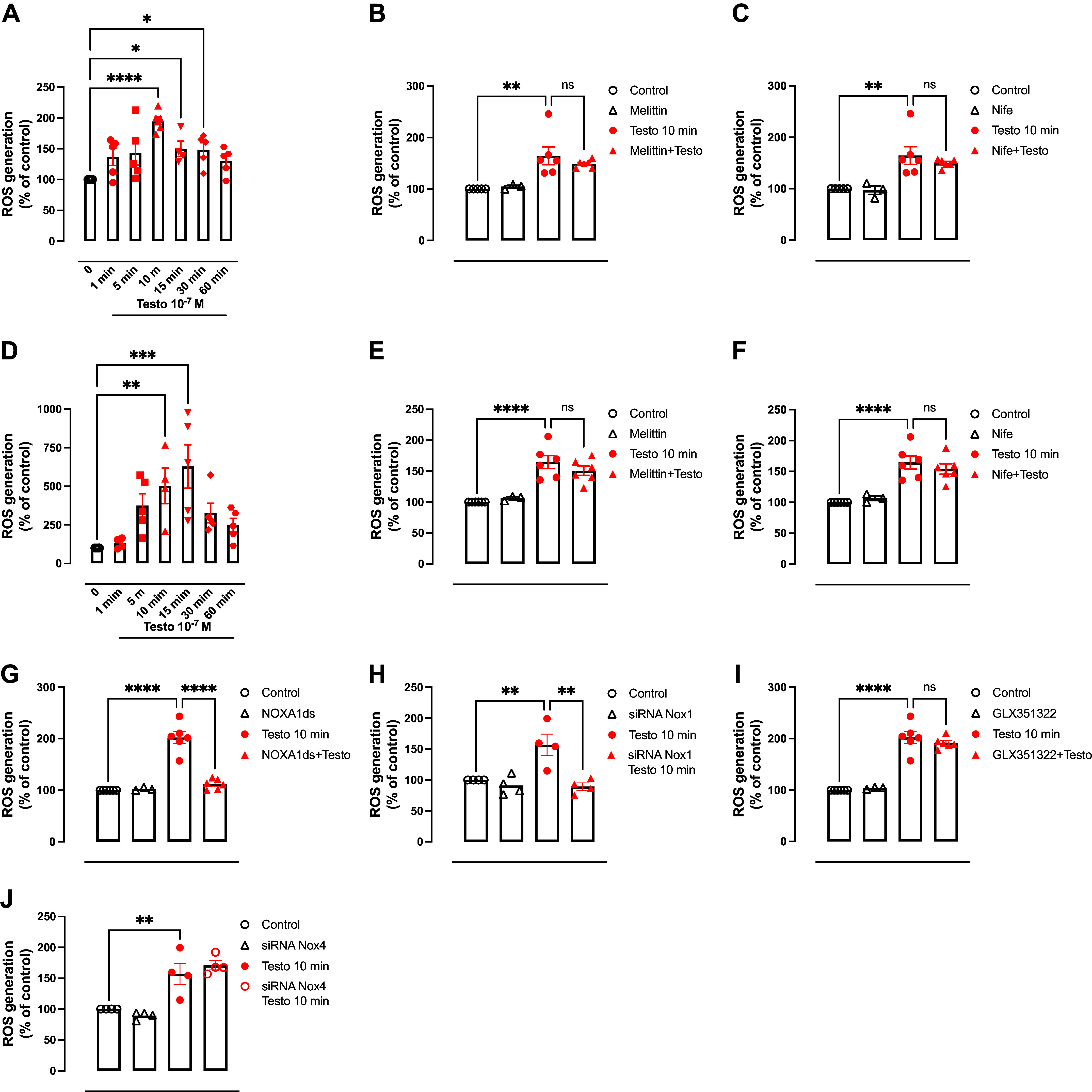
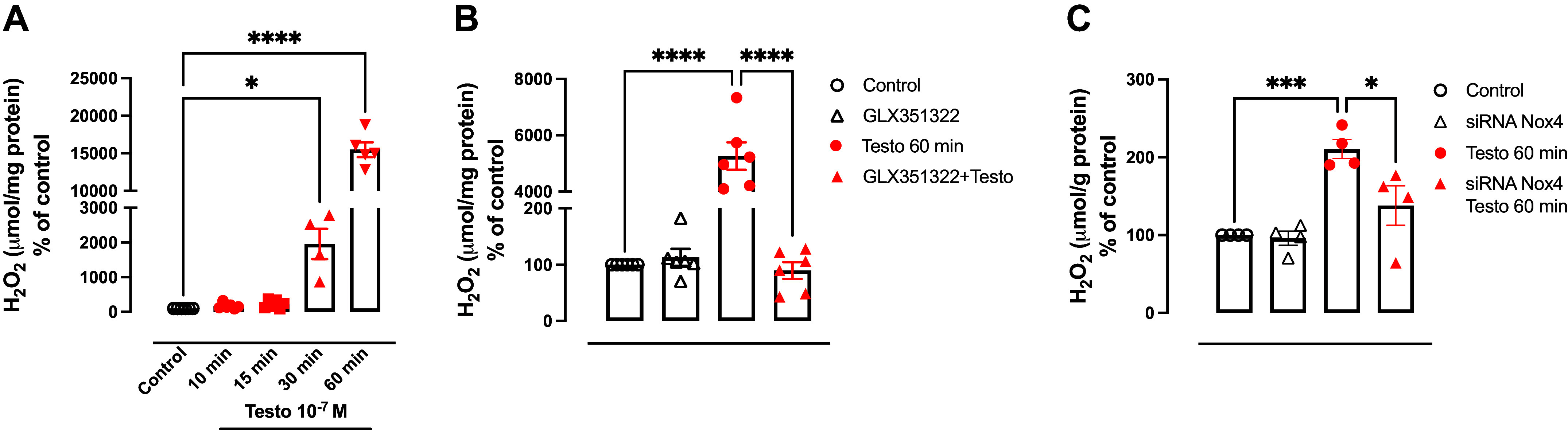
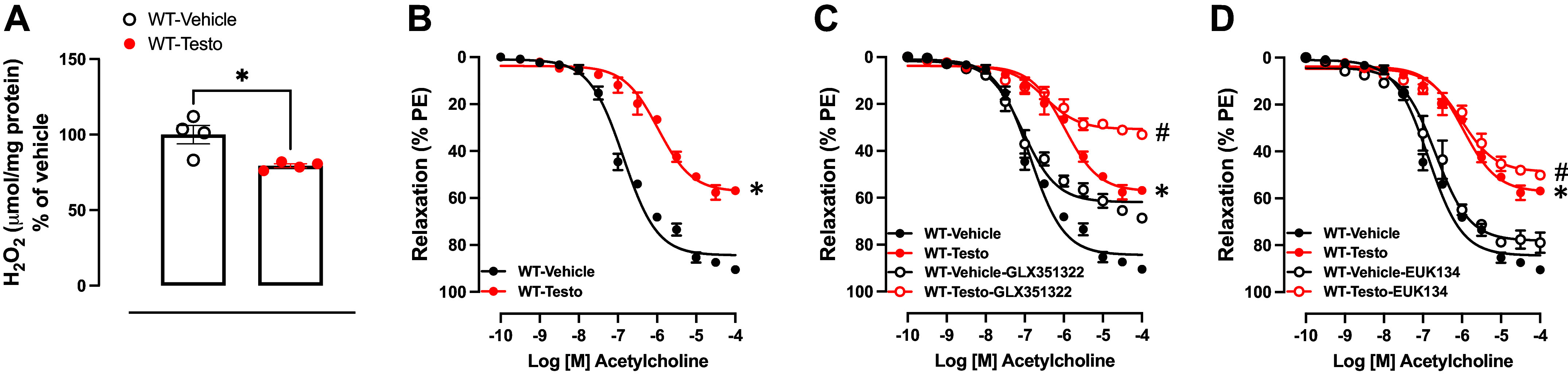
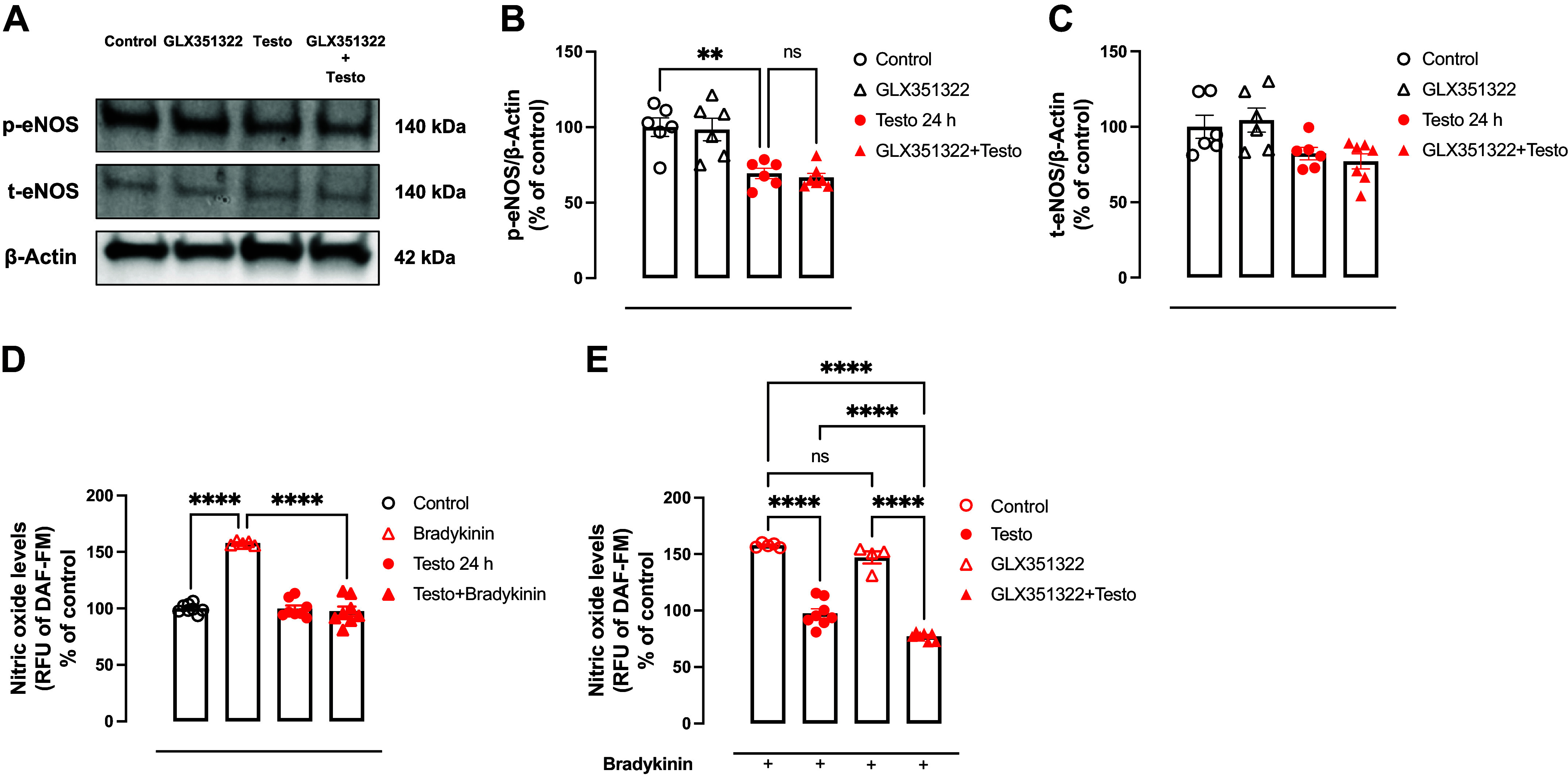
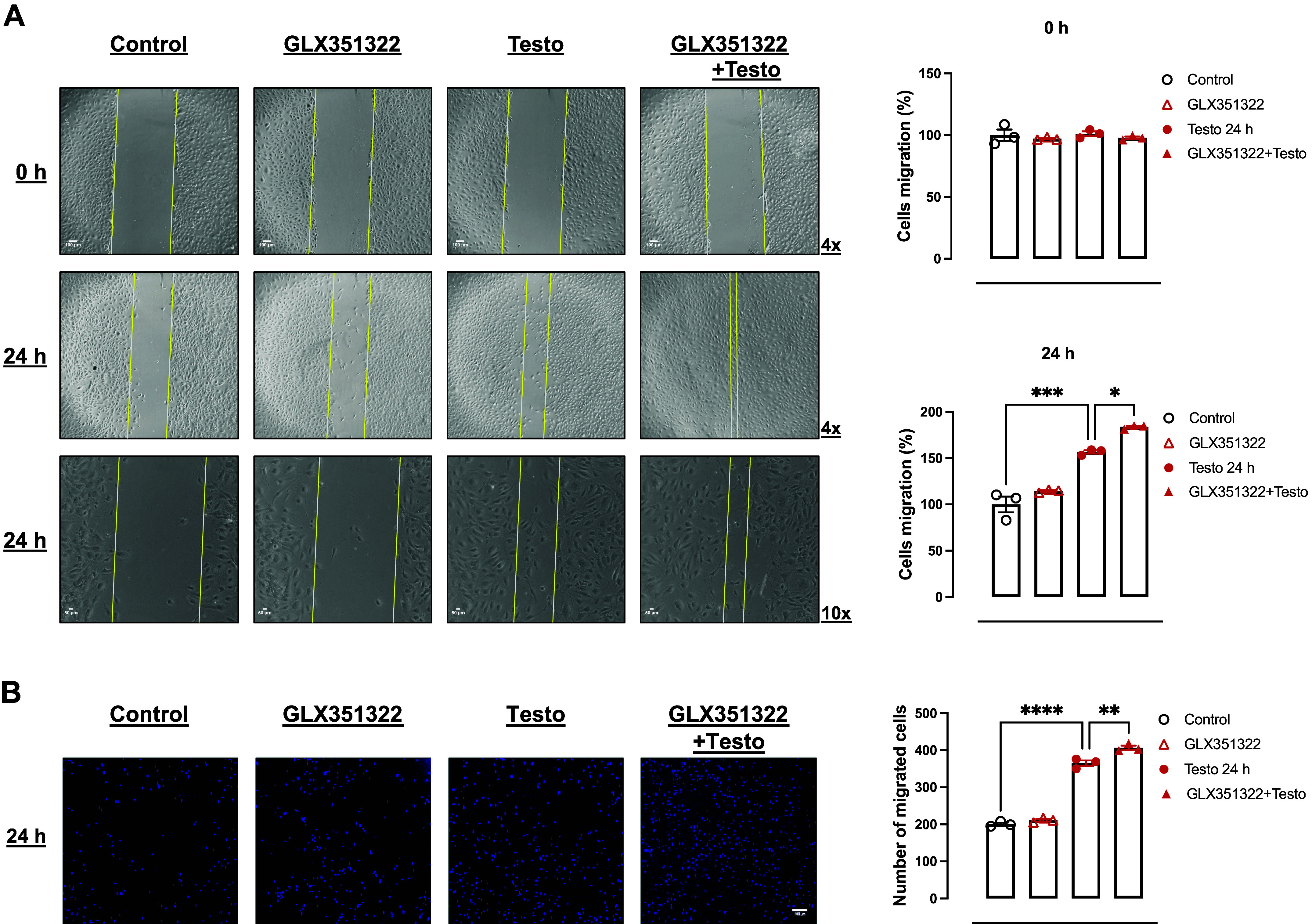
High levels of testosterone (Testo) are associated with cardiovascular risk by increasing reactive oxygen species (ROS) formation. NADPH oxidases (NOX) are the major source of ROS in the vasculature of cardiovascular diseases. NOX4 is a unique isotype, which produces hydrogen peroxide (H2O2), and its participation in cardiovascular biology is controversial. So far, it is unclear whether NOX4 protects from Testo-induced endothelial injury. Thus, we hypothesized that supraphysiological levels of Testo induce endothelial NOX4 expression to attenuate endothelial injury. Human mesenteric vascular endothelial cells (HMECs) and human umbilical vein endothelial cells (HUVEC) were treated with Testo (10-7 M) with or without a NOX4 inhibitor [GLX351322 (10-4 M)] or NOX4 siRNA. In vivo, 10-wk-old C57Bl/6J male mice were treated with Testo (10 mg/kg) for 30 days to study endothelial function. Testo increased mRNA and protein levels of NOX4 in HMECs and HUVECs. Testo increased superoxide anion (O2-) and H2O2 production, which were abolished by NOX1 and NOX4 inhibition, respectively. Testo also attenuated bradykinin-induced NO production, which was further impaired by NOX4 inhibition. In vivo, Testo decreased H2O2 production in aortic segments and triggered endothelial dysfunction [decreased relaxation to acetylcholine (ACh)], which was further impaired by GLX351322 and by a superoxide dismutase and catalase mimetic (EUK134). Finally, Testo led to a dysregulated endothelial cell migration, which was exacerbated by GLX351322. These data indicate that supraphysiological levels of Testo increase the endothelial expression and activity of NOX4 to counterbalance the deleterious effects caused by Testo in endothelial function.NEW & NOTEWORTHY By inducing ROS formation, high levels of testosterone play a major role in the pathogenesis of cardiovascular disease. NOXs are the major sources of ROS in the vasculature of cardiovascular diseases. Herein, we describe a novel compensatory mechanism by showing that NOX4 is a protective oxidant enzyme and counterbalances the deleterious effects of testosterone in endothelial cells by modulating hydrogen peroxide formation.
Keywords: NADPH oxidase; NOX4; endothelial dysfunction; oxidative stress; testosterone.
Conflict of interest statement
No conflicts of interest, financial or otherwise, are declared by the authors.
Figures
Similar articles
-
Breast cancer chemotherapy induces vascular dysfunction and hypertension through a NOX4-dependent mechanism.J Clin Invest. 2022 Jul 1;132(13):e149117. doi: 10.1172/JCI149117. J Clin Invest. 2022. PMID: 35617030 Free PMC article.
-
Aβ40 Improves Cerebrovascular Endothelial Function via NOX4-Dependent Hydrogen Peroxide Release.Int J Mol Sci. 2025 Jul 15;26(14):6759. doi: 10.3390/ijms26146759. Int J Mol Sci. 2025. PMID: 40725005 Free PMC article.
-
Overexpression of adipose tissue ERα enhances PVAT anticontractility via NOX4-derived H2O2 and is protective against high-fat diet-induced dysfunction.Am J Physiol Heart Circ Physiol. 2025 May 1;328(5):H1065-H1072. doi: 10.1152/ajpheart.00180.2025. Epub 2025 Mar 24. Am J Physiol Heart Circ Physiol. 2025. PMID: 40127093
-
Signs and symptoms to determine if a patient presenting in primary care or hospital outpatient settings has COVID-19.Cochrane Database Syst Rev. 2022 May 20;5(5):CD013665. doi: 10.1002/14651858.CD013665.pub3. Cochrane Database Syst Rev. 2022. PMID: 35593186 Free PMC article.
-
Sertindole for schizophrenia.Cochrane Database Syst Rev. 2005 Jul 20;2005(3):CD001715. doi: 10.1002/14651858.CD001715.pub2. Cochrane Database Syst Rev. 2005. PMID: 16034864 Free PMC article.
Cited by
-
LDL-c/HDL-c Ratio and NADPH-Oxidase-2-Derived Oxidative Stress as Main Determinants of Microvascular Endothelial Function in Morbidly Obese Subjects.Antioxidants (Basel). 2024 Sep 20;13(9):1139. doi: 10.3390/antiox13091139. Antioxidants (Basel). 2024. PMID: 39334798 Free PMC article.
-
An exploratory study on the metagenomic and proteomic characterization of hypothyroidism in the first half of pregnancy and correlation with Th1/Th2 balance.Front Immunol. 2025 May 15;16:1500866. doi: 10.3389/fimmu.2025.1500866. eCollection 2025. Front Immunol. 2025. PMID: 40443669 Free PMC article.
-
The effect of transient sex hormone fluctuations on vascular endothelial function.Am J Physiol Heart Circ Physiol. 2025 Jul 1;329(1):H217-H232. doi: 10.1152/ajpheart.00174.2025. Epub 2025 Jun 3. Am J Physiol Heart Circ Physiol. 2025. PMID: 40459955 Free PMC article. Review.
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases