

Paternal microbiome perturbations impact offspring fitness
- PMID: 38693261
- PMCID: PMC11096121
- DOI: 10.1038/s41586-024-07336-w
Paternal microbiome perturbations impact offspring fitness
Abstract
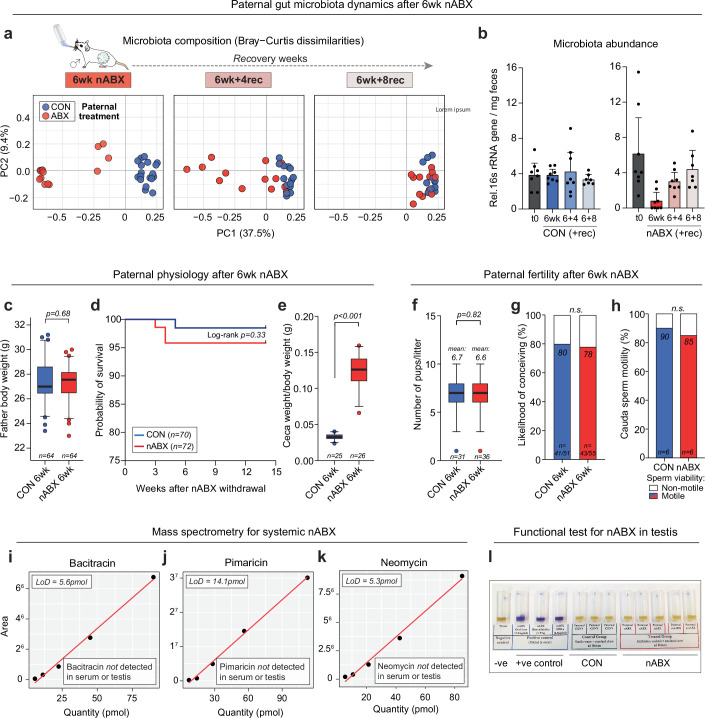
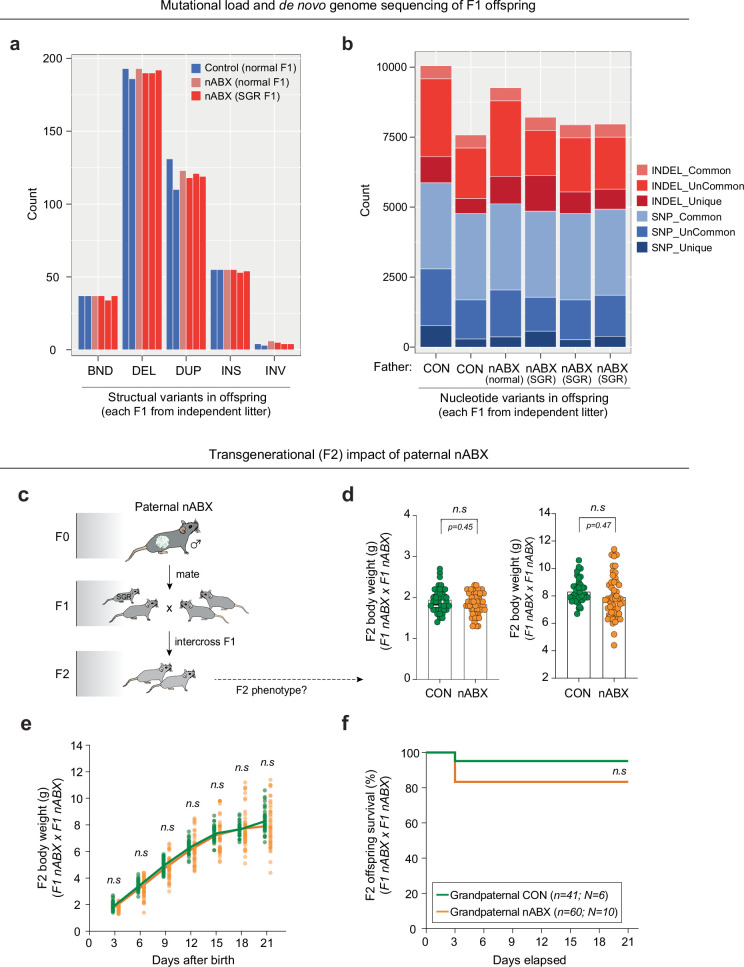
The gut microbiota operates at the interface of host-environment interactions to influence human homoeostasis and metabolic networks1-4. Environmental factors that unbalance gut microbial ecosystems can therefore shape physiological and disease-associated responses across somatic tissues5-9. However, the systemic impact of the gut microbiome on the germline-and consequently on the F1 offspring it gives rise to-is unexplored10. Here we show that the gut microbiota act as a key interface between paternal preconception environment and intergenerational health in mice. Perturbations to the gut microbiota of prospective fathers increase the probability of their offspring presenting with low birth weight, severe growth restriction and premature mortality. Transmission of disease risk occurs via the germline and is provoked by pervasive gut microbiome perturbations, including non-absorbable antibiotics or osmotic laxatives, but is rescued by restoring the paternal microbiota before conception. This effect is linked with a dynamic response to induced dysbiosis in the male reproductive system, including impaired leptin signalling, altered testicular metabolite profiles and remapped small RNA payloads in sperm. As a result, dysbiotic fathers trigger an elevated risk of in utero placental insufficiency, revealing a placental origin of mammalian intergenerational effects. Our study defines a regulatory 'gut-germline axis' in males, which is sensitive to environmental exposures and programmes offspring fitness through impacting placenta function.
© 2024. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures














Comment in
-
Paternal microbiome perturbations affect offspring outcomes.Nat Rev Urol. 2025 Jan;22(1):6-7. doi: 10.1038/s41585-024-00921-8. Nat Rev Urol. 2025. PMID: 39060488 Free PMC article. No abstract available.
References
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials
