

Genomes of multicellular algal sisters to land plants illuminate signaling network evolution
- PMID: 38693345
- PMCID: PMC11096116
- DOI: 10.1038/s41588-024-01737-3
Genomes of multicellular algal sisters to land plants illuminate signaling network evolution
Abstract
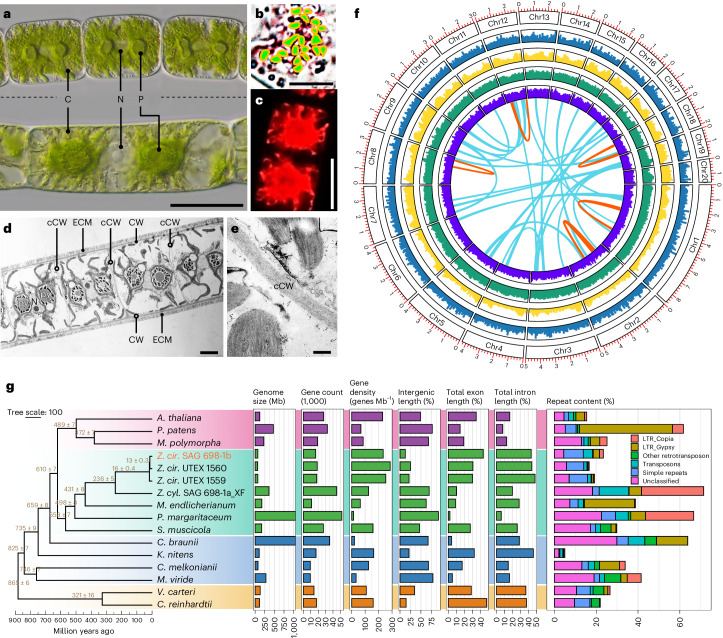
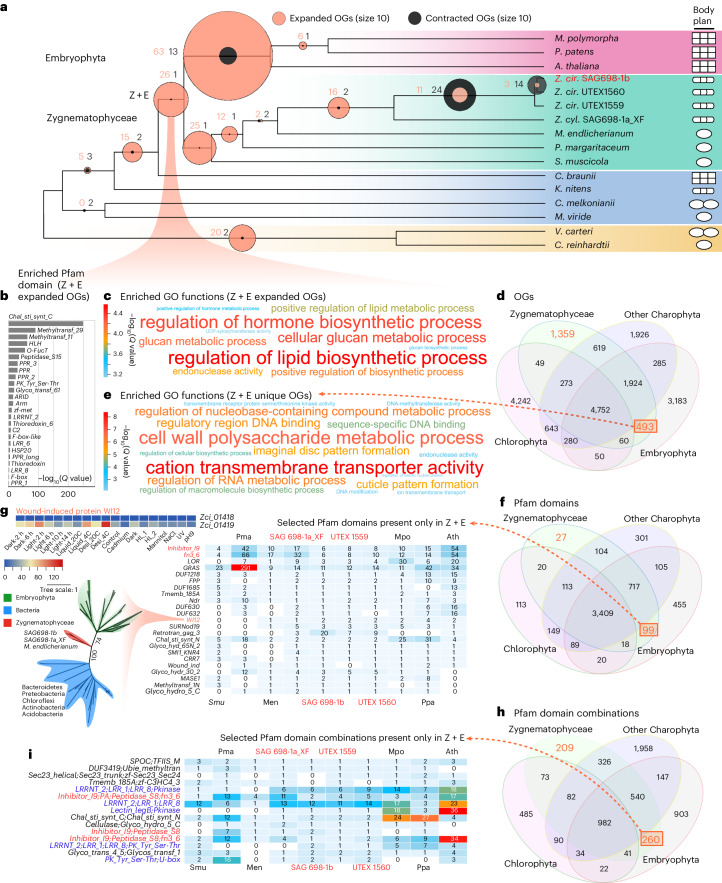
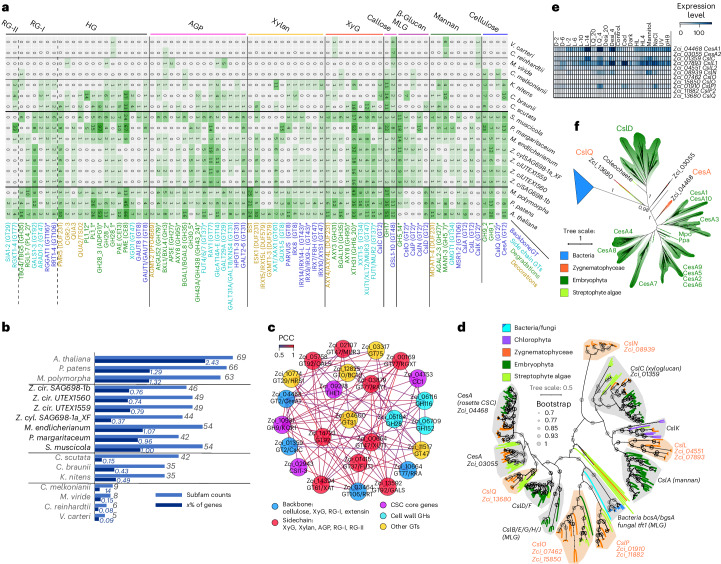
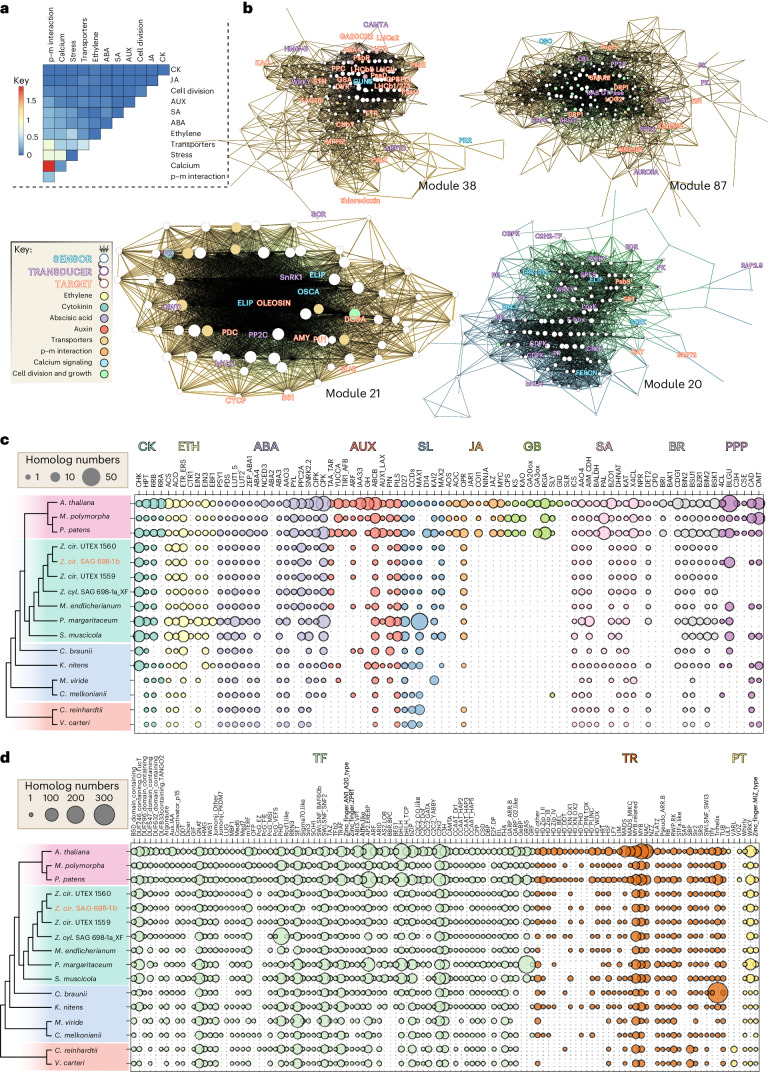
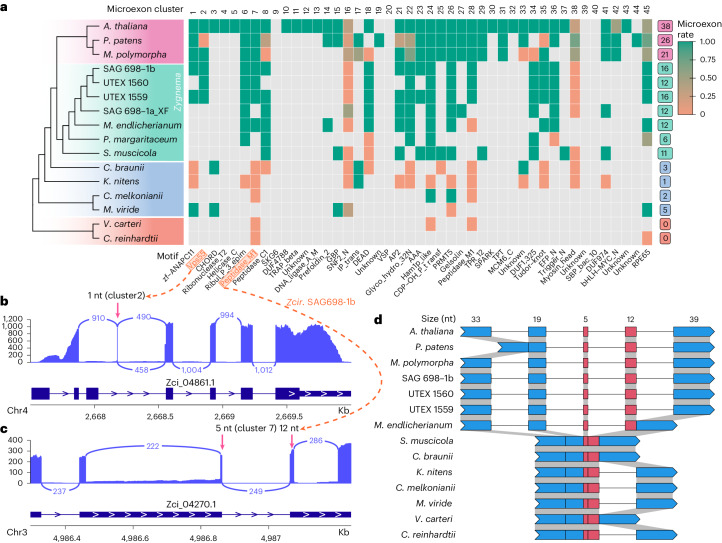
Zygnematophyceae are the algal sisters of land plants. Here we sequenced four genomes of filamentous Zygnematophyceae, including chromosome-scale assemblies for three strains of Zygnema circumcarinatum. We inferred traits in the ancestor of Zygnematophyceae and land plants that might have ushered in the conquest of land by plants: expanded genes for signaling cascades, environmental response, and multicellular growth. Zygnematophyceae and land plants share all the major enzymes for cell wall synthesis and remodifications, and gene gains shaped this toolkit. Co-expression network analyses uncover gene cohorts that unite environmental signaling with multicellular developmental programs. Our data shed light on a molecular chassis that balances environmental response and growth modulation across more than 600 million years of streptophyte evolution.
© 2024. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures

Update of
-
Chromosome-level genomes of multicellular algal sisters to land plants illuminate signaling network evolution.bioRxiv [Preprint]. 2023 Feb 1:2023.01.31.526407. doi: 10.1101/2023.01.31.526407. bioRxiv. 2023. Update in: Nat Genet. 2024 May;56(5):1018-1031. doi: 10.1038/s41588-024-01737-3. PMID: 36778228 Free PMC article. Updated. Preprint.
Comment in
-
Two steps beyond.Nat Plants. 2024 May;10(5):695. doi: 10.1038/s41477-024-01719-7. Nat Plants. 2024. PMID: 38773272 No abstract available.
References
-
- Guiry MD. AlgaeBase: a global database for algae. Curr. Sci. 2021;121:10–11.
MeSH terms
Grants and funding
- R01GM140370/Foundation for the National Institutes of Health (Foundation for the National Institutes of Health, Inc.)
- RE 1697/16-1/Deutsche Forschungsgemeinschaft (German Research Foundation)
- RE 1697/18-1/Deutsche Forschungsgemeinschaft (German Research Foundation)
- 1933521/National Science Foundation (NSF)
- R21AI171952/Foundation for the National Institutes of Health (Foundation for the National Institutes of Health, Inc.)
- DE-AC02-05CH11231/U.S. Department of Energy (DOE)
- 852725/EC | EU Framework Programme for Research and Innovation H2020 | H2020 Priority Excellent Science | H2020 European Research Council (H2020 Excellent Science - European Research Council)
- R01 GM140370/GM/NIGMS NIH HHS/United States
- R21 AI171952/AI/NIAID NIH HHS/United States
- P34181-B/Austrian Science Fund (Fonds zur Förderung der Wissenschaftlichen Forschung)
- 58-8042-9-089/United States Department of Agriculture | Agricultural Research Service (USDA Agricultural Research Service)
- ANR-10-LABX-41/LABoratoires d'EXcellence ARCANE (Labex ARCANE)
- 410739858/Deutsche Forschungsgemeinschaft (German Research Foundation)
- TH417/12-1/Deutsche Forschungsgemeinschaft (German Research Foundation)
- 440231723/Deutsche Forschungsgemeinschaft (German Research Foundation)
- OPP1172165/Bill and Melinda Gates Foundation (Bill & Melinda Gates Foundation)
- 101001675/EC | EU Framework Programme for Research and Innovation H2020 | H2020 Priority Excellent Science | H2020 European Research Council (H2020 Excellent Science - European Research Council)
- 440540015/Deutsche Forschungsgemeinschaft (German Research Foundation)
