

Congenital myasthenic syndromes in adults: clinical features, diagnosis and long-term prognosis
- PMID: 38696726
- PMCID: PMC11531845
- DOI: 10.1093/brain/awae124
Congenital myasthenic syndromes in adults: clinical features, diagnosis and long-term prognosis
Abstract
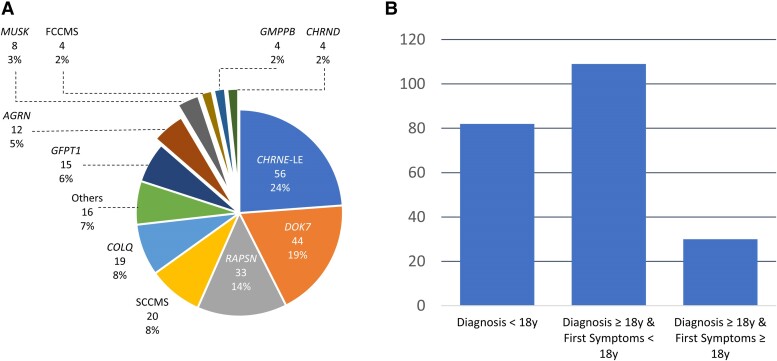
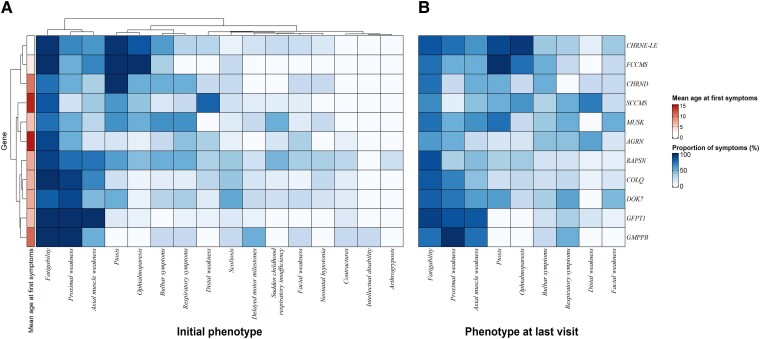
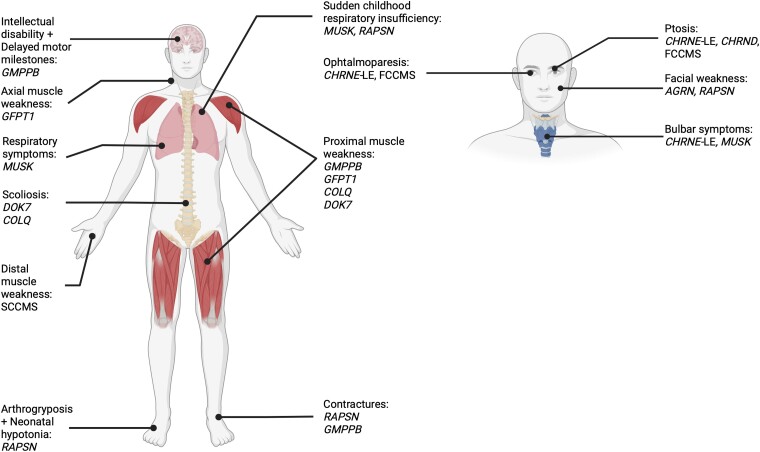
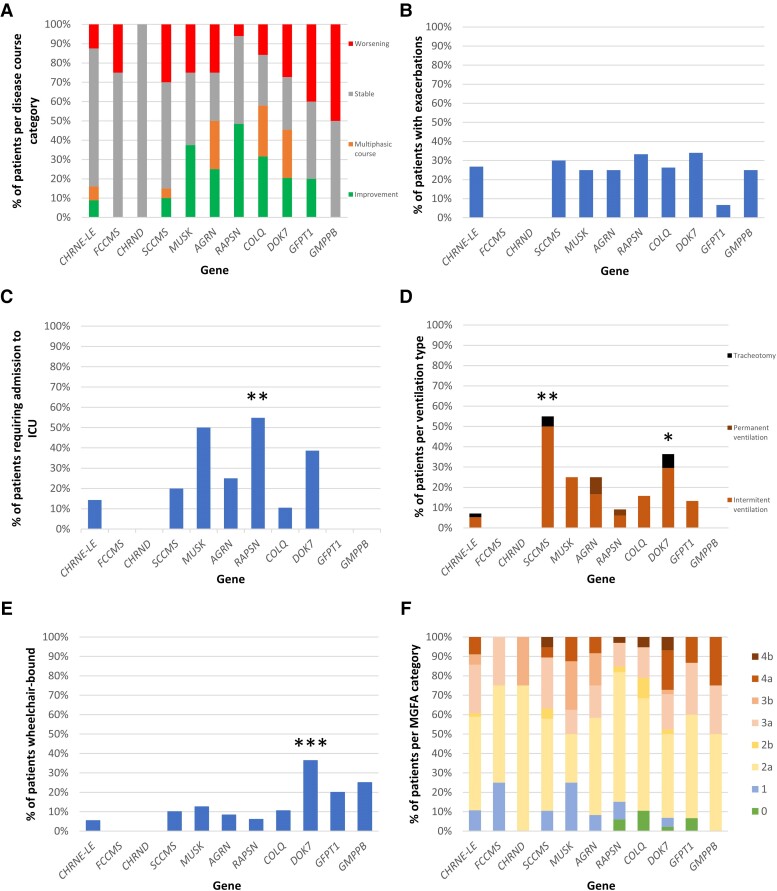
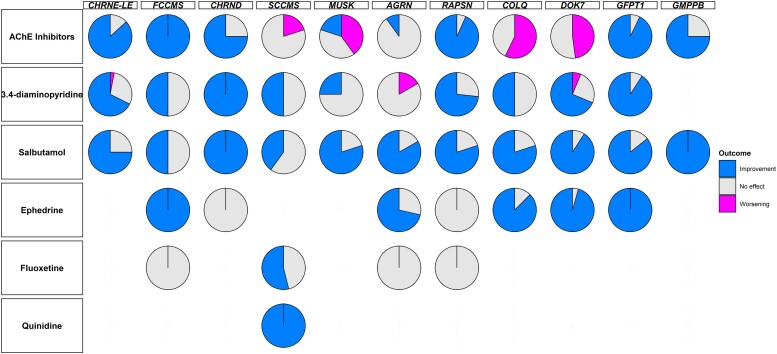
Congenital myasthenic syndromes (CMS) are clinically and genetically heterogeneous diseases caused by mutations affecting neuromuscular transmission. Even if the first symptoms mainly occur during childhood, adult neurologists must confront this challenging diagnosis and manage these patients throughout their adulthood. However, long-term follow-up data from large cohorts of CMS patients are lacking, and the long-term prognosis of these patients is largely unknown. We report the clinical features, diagnostic difficulties, and long-term prognosis of a French nationwide cohort of 235 adult patients with genetically confirmed CMS followed in 23 specialized neuromuscular centres. Data were retrospectively analysed. Of the 235 patients, 123 were female (52.3%). The diagnosis was made in adulthood in 139 patients, 110 of whom presented their first symptoms before the age of 18. Mean follow-up time between first symptoms and last visit was 34 years [standard deviation (SD) = 15.1]. Pathogenic variants were found in 19 disease-related genes. CHRNE-low expressor variants were the most common (23.8%), followed by variants in DOK7 (18.7%) and RAPSN (14%). Genotypes were clustered into four groups according to the initial presentation: ocular group (CHRNE-LE, CHRND, FCCMS), distal group (SCCMS), limb-girdle group (RAPSN, COLQ, DOK7, GMPPB, GFPT1), and a variable-phenotype group (MUSK, AGRN). The phenotypical features of CMS did not change throughout life. Only four genotypes had a proportion of patients requiring intensive care unit admission that exceeded 20%: RAPSN (54.8%), MUSK (50%), DOK7 (38.6%) and AGRN (25.0%). In RAPSN and MUSK patients most ICU admissions occurred before age 18 years and in DOK7 and AGRN patients at or after 18 years of age. Different patterns of disease course (stability, improvement and progressive worsening) may succeed one another in the same patient throughout life, particularly in AGRN, DOK7 and COLQ. At the last visit, 55% of SCCMS and 36.3% of DOK7 patients required ventilation; 36.3% of DOK7 patients, 25% of GMPPB patients and 20% of GFPT1 patients were wheelchair-bound; most of the patients who were both wheelchair-bound and ventilated were DOK7 patients. Six patients died in this cohort. The positive impact of therapy was striking, even in severely affected patients. In conclusion, even if motor and/or respiratory deterioration could occur in patients with initially moderate disease, particularly in DOK7, SCCMS and GFPT1 patients, the long-term prognosis for most CMS patients was favourable, with neither ventilation nor wheelchair needed at last visit. CHRNE-LE patients did not worsen during adulthood and RAPSN patients, often severely affected in early childhood, subsequently improved.
Keywords: CMS; genetic; myasthenia; neuromuscular junction.
© The Author(s) 2024. Published by Oxford University Press on behalf of the Guarantors of Brain.
Conflict of interest statement
The authors report no competing interests.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Miscellaneous
