

This is a preprint.
The peripheral epigenome predicts white matter volume contingent on developmental stage: An ECHO study
- PMID: 38699338
- PMCID: PMC11065062
- DOI: 10.21203/rs.3.rs-4139933/v1
The peripheral epigenome predicts white matter volume contingent on developmental stage: An ECHO study
Abstract
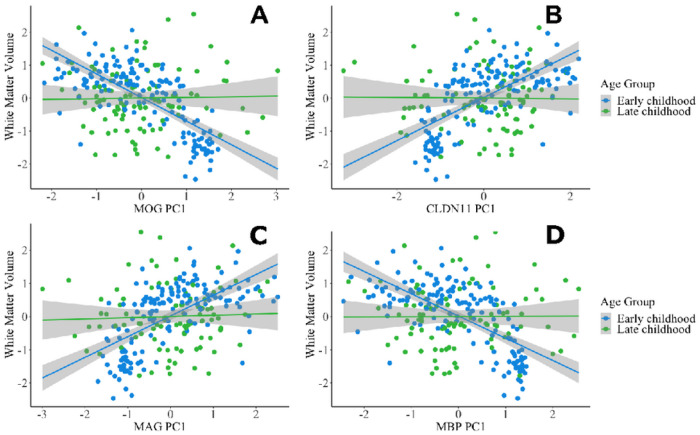
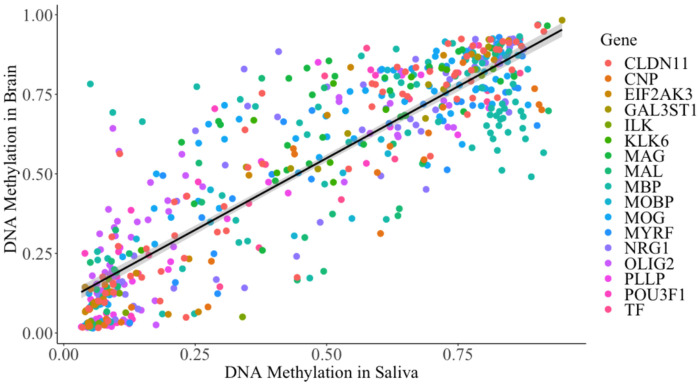
Epigenetic processes, including DNA methylation, are emerging as key areas of interest for their potential roles as biomarkers and contributors to the risk of neurodevelopmental, psychiatric, and other brain-based disorders. Despite this growing focus, there remains a notable gap in our understanding of how DNA methylation correlates with individual variations in brain function and structure. Additionally, the dynamics of these relationships during developmental periods, which are critical windows during which many disorders first appear, are still largely unexplored. The current study extends the field by examining if peripheral DNA methylation of myelination-related genes predicts white matter volume in a healthy pediatric population [N = 250; females = 113; age range 2 months-14 years; Mage = 5.14, SDage = 3.60]. We assessed if DNA methylation of 17 myelin-related genes predict white matter volume and if age moderates these relationships. Results highlight low variability in myelin-related epigenetic variance at birth, which rapidly increases non-linearly with age, and that DNA methylation, measured at both the level of a CpG site or gene, is highly predictive of white matter volume, in early childhood but not late childhood. These novel findings propel the field forward by establishing that DNA methylation of myelin-related genes from a peripheral tissue is a predictive marker of white matter volume in children and is influenced by developmental stage. The research underscores the significance of peripheral epigenetic patterns as a proxy for investigating the effects of environmental factors, behaviors, and disorders associated with white matter.
Conflict of interest statement
Additional Declarations: No competing interests reported. Competing interests The authors have no conflict of interest to disclose. Supplementary Files This is a list of supplementary files associated with this preprint. Click to download. • SupplementalInformation.docx
Figures

References
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
