

Genetically Engineered CLDN18.2 CAR-T Cells Expressing Synthetic PD1/CD28 Fusion Receptors Produced Using a Lentiviral Vector
- PMID: 38700775
- PMCID: PMC11303488
- DOI: 10.1007/s12275-024-00133-0
Genetically Engineered CLDN18.2 CAR-T Cells Expressing Synthetic PD1/CD28 Fusion Receptors Produced Using a Lentiviral Vector
Abstract
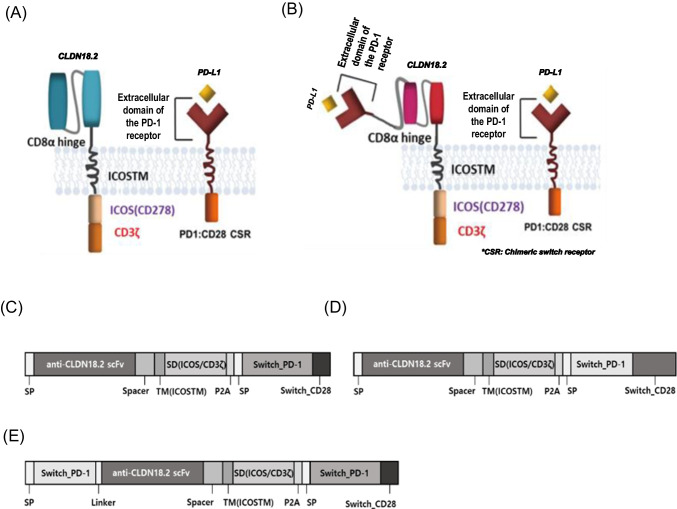
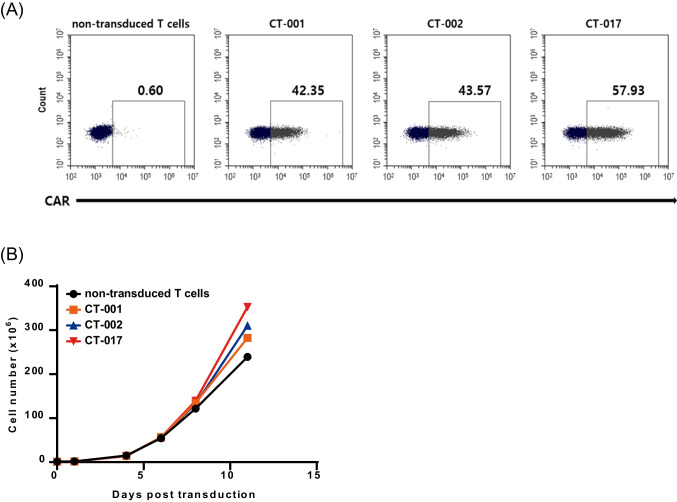
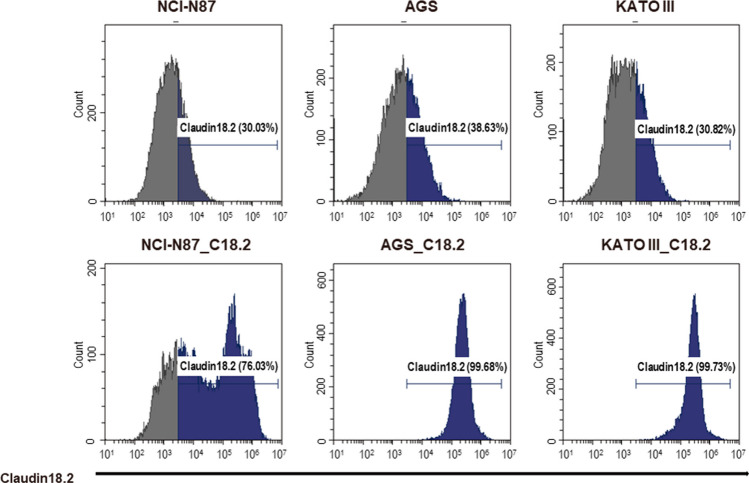
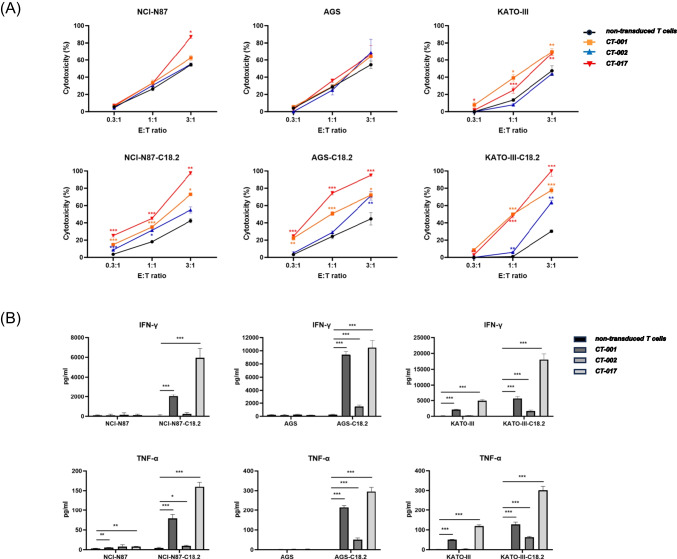
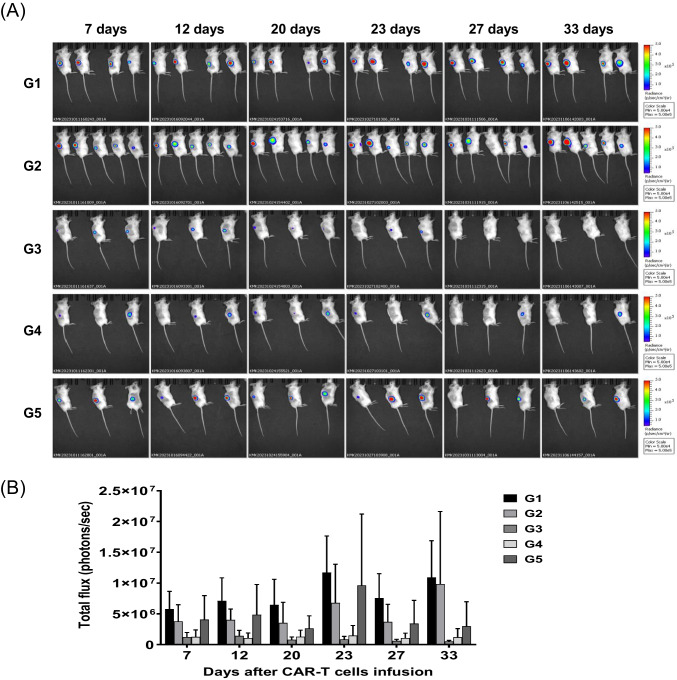
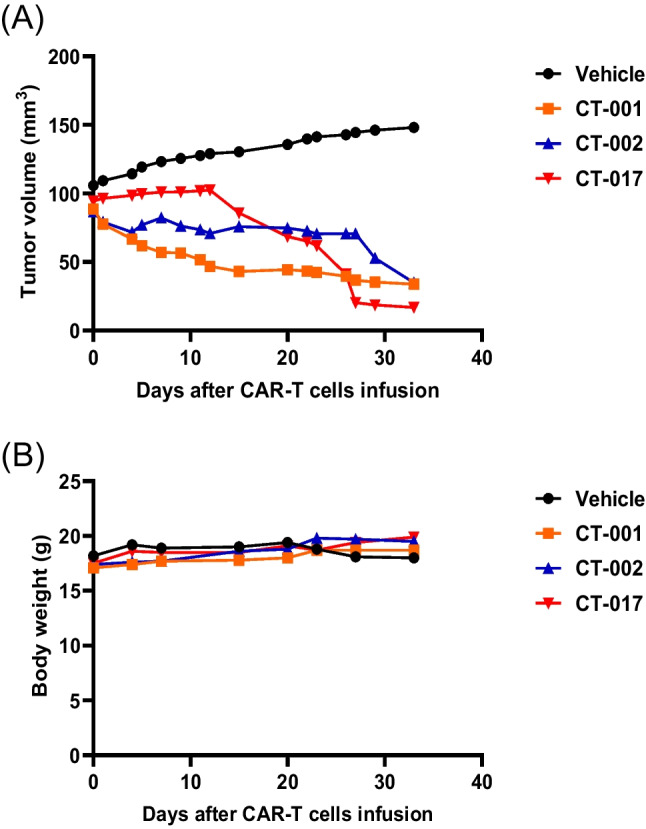
This study aimed to develop synthetic Claudin18.2 (CLDN18.2) chimeric antigen receptor (CAR)-T (CAR-T) cells as a treatment for advanced gastric cancer using lentiviral vector genetic engineering technology that targets the CLDN18.2 antigen and simultaneously overcomes the immunosuppressive environment caused by programmed cell death protein 1 (PD-1). Synthetic CAR T cells are a promising approach in cancer immunotherapy but face many challenges in solid tumors. One of the major problems is immunosuppression caused by PD-1. CLDN18.2, a gastric-specific membrane protein, is considered a potential therapeutic target for gastric and other cancers. In our study, CLDN18.2 CAR was a second-generation CAR with inducible T-cell costimulatory (CD278), and CLDN18.2-PD1/CD28 CAR was a third-generation CAR, wherein the synthetic PD1/CD28 chimeric-switch receptor (CSR) was added to the second-generation CAR. In vitro, we detected the secretion levels of different cytokines and the killing ability of CAR-T cells. We found that the secretion of cytokines such as interferon-gamma (IFN-γ) and tumor necrosis factor-alpha (TNF-α) secreted by three types of CAR-T cells was increased, and the killing ability against CLDN18.2-positive GC cells was enhanced. In vivo, we established a xenograft GC model and observed the antitumor effects and off-target toxicity of CAR-T cells. These results support that synthetic anti-CLDN18.2 CAR-T cells have antitumor effect and anti-CLDN18.2-PD1/CD28 CAR could provide a promising design strategy to improve the efficacy of CAR-T cells in advanced gastric cancer.
Keywords: CAR-T; Claudin18.2; Gastric cancer; Lentiviral vector; Synthetic PD1/CD28 receptor.
© 2024. The Author(s).
Conflict of interest statement
The authors have no financial conflicts of interest to declare.
Figures
References
-
- Alzubi, J., Dettmer-Monaco, V., Kuehle, J., Thorausch, N., Seidl, M., Taromi, S., Schamel, W., Zeiser, R., Abken, H., Cathomen, T., et al. (2020). PSMA-directed CAR T cells combined with low-dose docetaxel treatment induce tumor regression in a prostate cancer xenograft model. Molecular Therapy Oncolytics,18, 226–235. 10.1016/j.omto.2020.06.014 - DOI - PMC - PubMed
-
- Bates, P. D., Rakhmilevich, A. L., Cho, M. M., Bouchlaka, M. N., Rao, S. L., Hales, J. M., Orentas, R. J., Fry, T. J., Gilles, S. D., Sondel, P. M., et al. (2021). combining immunocytokine and ex vivo activated NK cells as a platform for enhancing graft-versus-tumor effects against GD2+ murine neuroblastoma. Frontiers in Immunology,12, 668307. 10.3389/fimmu.2021.668307 - DOI - PMC - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
