

Motive and opportunity: MYC rearrangements in high-grade B-cell lymphoma with MYC and BCL2 rearrangements (an LLMPP study)
- PMID: 38701426
- PMCID: PMC11307266
- DOI: 10.1182/blood.2024024251
Motive and opportunity: MYC rearrangements in high-grade B-cell lymphoma with MYC and BCL2 rearrangements (an LLMPP study)
Abstract
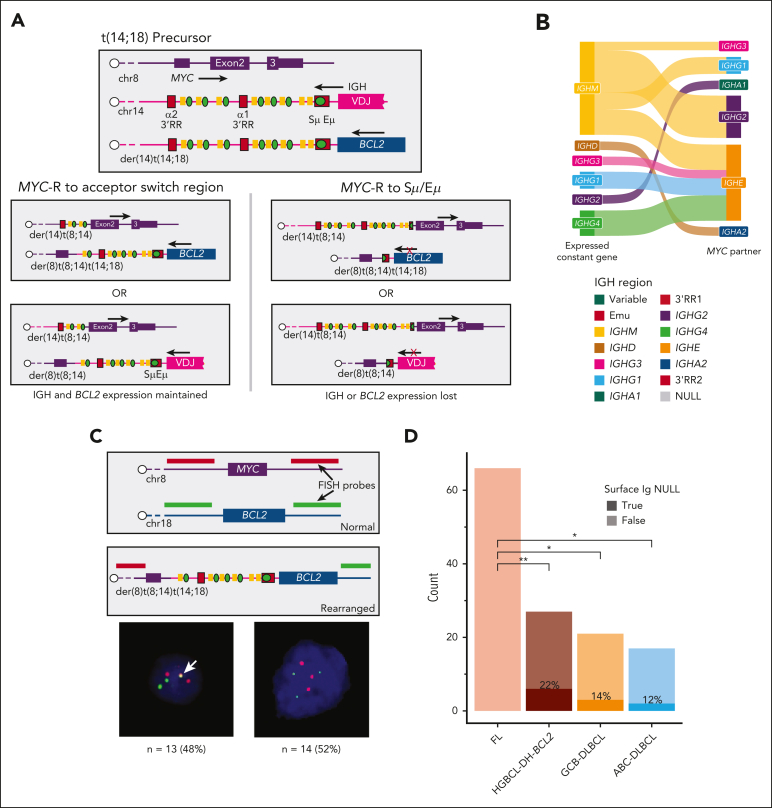
Rearrangements that place the oncogenes MYC, BCL2, or BCL6 adjacent to superenhancers are common in mature B-cell lymphomas. Lymphomas with diffuse large B-cell lymphoma (DLBCL) or high-grade morphology with both MYC and BCL2 rearrangements are classified as high-grade B-cell lymphoma with MYC and BCL2 rearrangements ("double hit"; HGBCL-DH-BCL2) and are associated with aggressive disease and poor outcomes. Although it is established that MYC rearrangements involving immunoglobulin (IG) loci are associated with inferior outcomes relative to those involving other non-IG superenhancers, the frequency of and mechanisms driving IG vs non-IG MYC rearrangements have not been elucidated. Here, we used custom targeted capture and/or whole-genome sequencing to characterize oncogene rearrangements across 883 mature B-cell lymphomas including Burkitt lymphoma, follicular lymphoma, DLBCL, and HGBCL-DH-BCL2 tumors. We demonstrate that, although BCL2 rearrangement topology is consistent across entities, HGBCL-DH-BCL2 have distinct MYC rearrangement architecture relative to tumors with single MYC rearrangements or with both MYC and BCL6 rearrangements (HGBCL-DH-BCL6), including both a higher frequency of non-IG rearrangements and different architecture of MYC::IGH rearrangements. The distinct MYC rearrangement patterns in HGBCL-DH-BCL2 occur on the background of high levels of somatic hypermutation across MYC partner loci in HGBCL-DH-BCL2, creating more opportunity to form these rearrangements. Furthermore, because 1 IGH allele is already disrupted by the existing BCL2 rearrangement, the MYC rearrangement architecture in HGBCL-DH-BCL2 likely reflects selective pressure to preserve both BCL2 and B-cell receptor expression. These data provide new mechanistic explanations for the distinct patterns of MYC rearrangements observed across different lymphoma entities.
Conflict of interest statement
Conflict-of-interest disclosure: D.W.S., R.D.M., L.M.R., G.W., L.M.S., E.C., T.C.G., J.R.C., K.F., E.S.J., A.R., W.C.C., G.I., J.D., and D.D.W. are named inventors on patents for the use of gene expression to subtype aggressive B-cell lymphomas, one of which is licensed to NanoString Technologies. C.S. reports consultancy fees from AbbVie, Bayer, and Seattle Genetics; and research funds from Trillium Therapeutics, Bristol Myers Squibb (BMS), and Epizyme. L.H.S. reports consulting fees/honoraria from AbbVie, Amgen, AstraZeneca, BeiGene, BMS/Celgene, GenMab, Kite/Gilead, Incyte, Janssen, Merck, Seagen, and Roche/Genentech; and research funding from Roche/Genentech and Teva. K.J.S. reports honoraria/consulting fees from BMS, Merck, Seagen, Janssen, and AbbVie; steering committee fees from AstraZeneca; research funding from BMS and Roche; and the data safety monitoring committee for Regeneron. D.W.S. reports consulting fees/honoraria from AbbVie, AstraZeneca, GenMab, Incyte, Roche/Genentech, and Veracyte and research funding from Roche/Genentech. The remaining authors declare no competing financial interests.
Figures








Comment in
-
MYC translocation architecture in B-NHL.Blood. 2024 Aug 1;144(5):469-471. doi: 10.1182/blood.2024024945. Blood. 2024. PMID: 39088230 No abstract available.
References
-
- Ott G, Rosenwald A, Campo E. Understanding MYC-driven aggressive B-cell lymphomas: pathogenesis and classification. Blood. 2013;122(24):3884–3891. - PubMed
-
- Carbone A, Roulland S, Gloghini A, et al. Follicular lymphoma. Nat Rev Dis Primer. 2019;5(1):83. - PubMed
-
- Rosenwald A, Bens S, Advani R, et al. Prognostic significance of MYC rearrangement and translocation partner in diffuse large B-cell lymphoma: a study by the Lunenburg Lymphoma Biomarker Consortium. J Clin Oncol. 2019;37(35):3359–3368. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
