

A human neural crest model reveals the developmental impact of neuroblastoma-associated chromosomal aberrations
- PMID: 38702304
- PMCID: PMC11068915
- DOI: 10.1038/s41467-024-47945-7
A human neural crest model reveals the developmental impact of neuroblastoma-associated chromosomal aberrations
Abstract
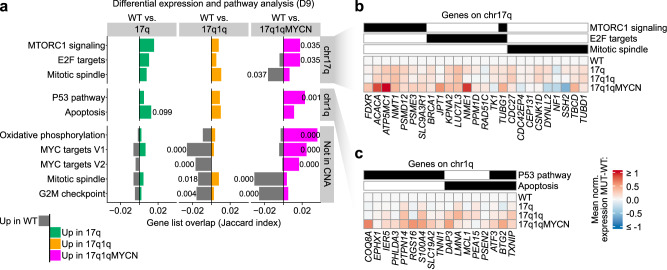
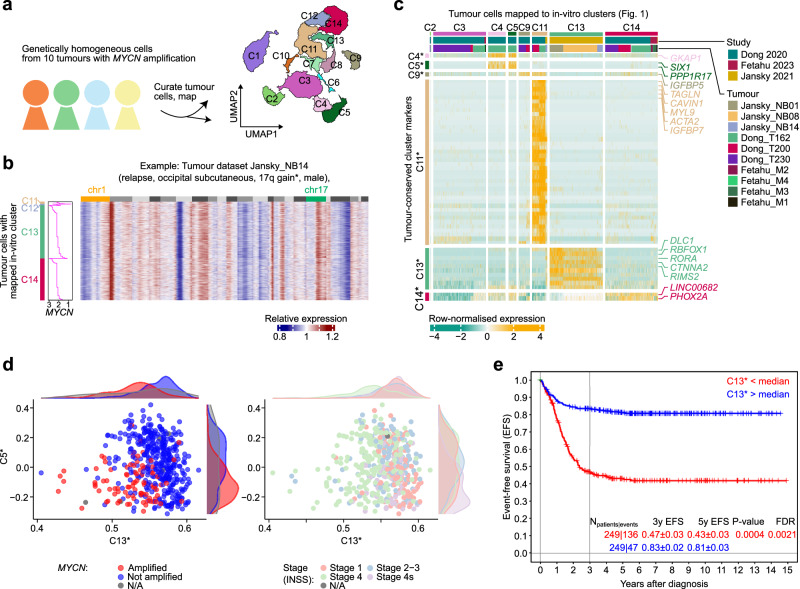
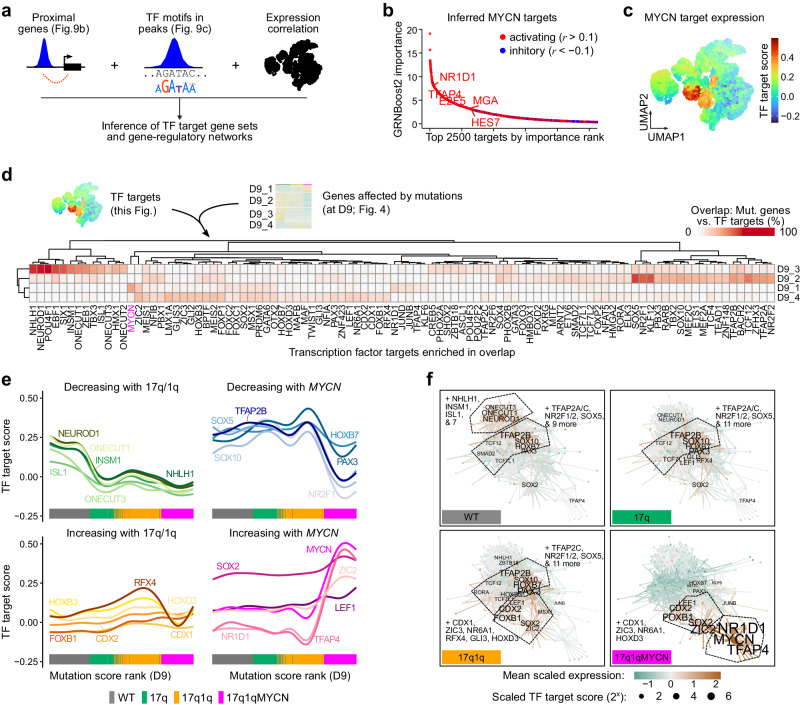
Early childhood tumours arise from transformed embryonic cells, which often carry large copy number alterations (CNA). However, it remains unclear how CNAs contribute to embryonic tumourigenesis due to a lack of suitable models. Here we employ female human embryonic stem cell (hESC) differentiation and single-cell transcriptome and epigenome analysis to assess the effects of chromosome 17q/1q gains, which are prevalent in the embryonal tumour neuroblastoma (NB). We show that CNAs impair the specification of trunk neural crest (NC) cells and their sympathoadrenal derivatives, the putative cells-of-origin of NB. This effect is exacerbated upon overexpression of MYCN, whose amplification co-occurs with CNAs in NB. Moreover, CNAs potentiate the pro-tumourigenic effects of MYCN and mutant NC cells resemble NB cells in tumours. These changes correlate with a stepwise aberration of developmental transcription factor networks. Together, our results sketch a mechanistic framework for the CNA-driven initiation of embryonal tumours.
© 2024. The Author(s).
Conflict of interest statement
P.W.A. is a member of the Scientific Advisory Board of TreeFrog Therapeutics and receives royalties from the Wistar Institute for antibody sales. All other authors declare no competing interests.
Figures







References
Publication types
MeSH terms
Substances
Grants and funding
- MR/R015724/1/RCUK | Medical Research Council (MRC)
- 20-17258/Alex's Lemonade Stand Foundation for Childhood Cancer (Alex's Lemonade Stand Foundation)
- P 35072/FWF_/Austrian Science Fund FWF/Austria
- 25905/Österreichischen Akademie der Wissenschaften (Austrian Academy of Sciences)
- 10.55776/P35841B/Austrian Science Fund (Fonds zur Förderung der Wissenschaftlichen Forschung)
- A28278/Cancer Research UK (CRUK)
- BB/P000444/1/RCUK | Biotechnology and Biological Sciences Research Council (BBSRC)
- MR/X000028/1/RCUK | Medical Research Council (MRC)
- TAI 732/FWF_/Austrian Science Fund FWF/Austria
- 10.55776/P35072/Austrian Science Fund (Fonds zur Förderung der Wissenschaftlichen Forschung)
- 10.55776/TAI732/Austrian Science Fund (Fonds zur Förderung der Wissenschaftlichen Forschung)
- 10.55776/TAI454/Austrian Science Fund (Fonds zur Förderung der Wissenschaftlichen Forschung)
- CCLGA 2020 19/Children's Cancer and Leukaemia Group (CCLG)
- BB/T007222/1/RCUK | Biotechnology and Biological Sciences Research Council (BBSRC)
- 10.55776/P32001/Austrian Science Fund (Fonds zur Förderung der Wissenschaftlichen Forschung)
- MR/X007979/1/RCUK | Medical Research Council (MRC)
- MR/V002163/1/RCUK | Medical Research Council (MRC)
- P 35841/FWF_/Austrian Science Fund FWF/Austria
- 10.55776/PAT1300223/Austrian Science Fund (Fonds zur Förderung der Wissenschaftlichen Forschung)
- 824070/EC | Horizon 2020 Framework Programme (EU Framework Programme for Research and Innovation H2020)
- LS18-111/Vienna Science and Technology Fund (Wiener Wissenschafts-, Forschungs- und Technologiefonds)
- CCLGA 2018 06/Children's Cancer and Leukaemia Group (CCLG)
- TAI 454/FWF_/Austrian Science Fund FWF/Austria
- 10.55776/P34832/Austrian Science Fund (Fonds zur Förderung der Wissenschaftlichen Forschung)
- R00CA197484/U.S. Department of Health & Human Services | National Institutes of Health (NIH)
- CCLGA 2019 28/Children's Cancer and Leukaemia Group (CCLG)
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
