

Systematic identification of structure-specific protein-protein interactions
- PMID: 38702390
- PMCID: PMC11148107
- DOI: 10.1038/s44320-024-00037-6
Systematic identification of structure-specific protein-protein interactions
Abstract
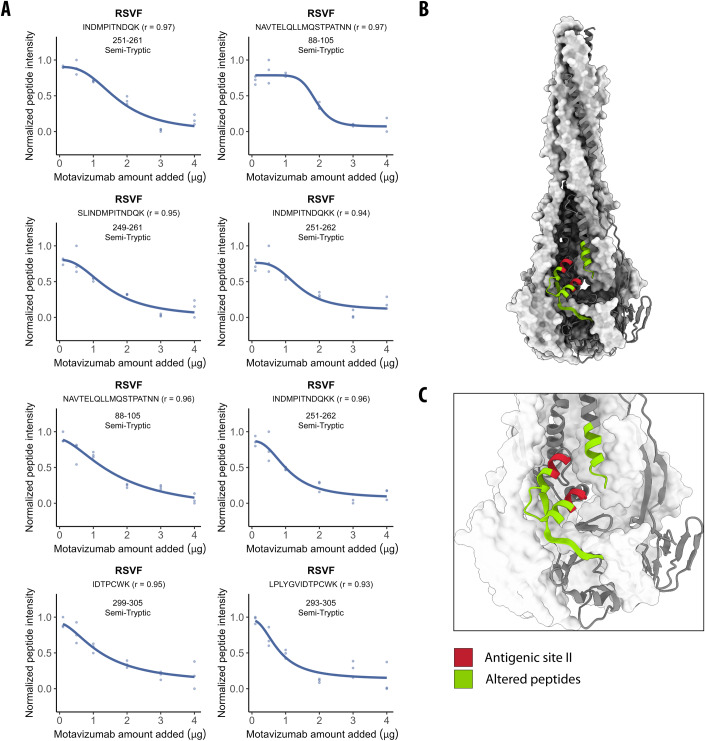
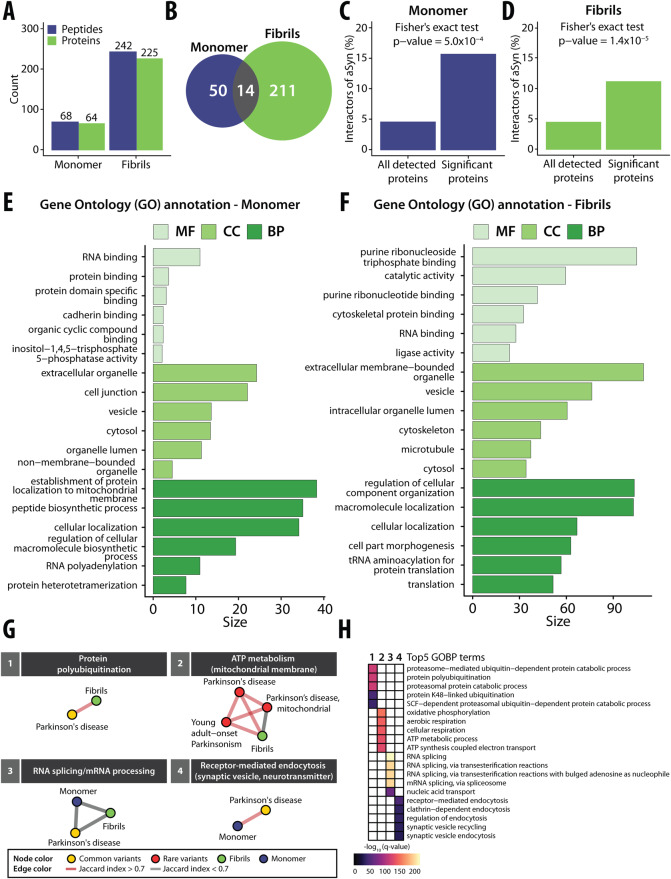
The physical interactome of a protein can be altered upon perturbation, modulating cell physiology and contributing to disease. Identifying interactome differences of normal and disease states of proteins could help understand disease mechanisms, but current methods do not pinpoint structure-specific PPIs and interaction interfaces proteome-wide. We used limited proteolysis-mass spectrometry (LiP-MS) to screen for structure-specific PPIs by probing for protease susceptibility changes of proteins in cellular extracts upon treatment with specific structural states of a protein. We first demonstrated that LiP-MS detects well-characterized PPIs, including antibody-target protein interactions and interactions with membrane proteins, and that it pinpoints interfaces, including epitopes. We then applied the approach to study conformation-specific interactors of the Parkinson's disease hallmark protein alpha-synuclein (aSyn). We identified known interactors of aSyn monomer and amyloid fibrils and provide a resource of novel putative conformation-specific aSyn interactors for validation in further studies. We also used our approach on GDP- and GTP-bound forms of two Rab GTPases, showing detection of differential candidate interactors of conformationally similar proteins. This approach is applicable to screen for structure-specific interactomes of any protein, including posttranslationally modified and unmodified, or metabolite-bound and unbound protein states.
Keywords: Limited Proteolysis; Mass Spectrometry; Protein–protein Interactions; Structural Proteomics; Structure-Specific Interactions.
© 2024. The Author(s).
Conflict of interest statement
PP is an inventor of a patent licensed by Biognosys AG that covers the LiP–MS method used in this manuscript and a member of the scientific advisory board of Biognosys AG. The remaining authors declare no competing interests. Pedro Beltrao is a member of the Advisory Editorial Board of Molecular Systems Biology. This has no bearing on the editorial consideration of this article for publication.
Figures



References
-
- Afitska K, Priss A, Yushchenko DA, Shvadchak VV. Structural optimization of inhibitors of α-synuclein fibril growth: affinity to the fibril end as a crucial factor. J Mol Biol. 2020;432:967–977. - PubMed
-
- Antoniou N, Prodromidou K, Kouroupi G, Boumpoureka I, Samiotaki M, Panayotou G, Xilouri M, Kloukina I, Stefanis L, Grailhe R, et al. High content screening and proteomic analysis identify a kinase inhibitor that rescues pathological phenotypes in a patient-derived model of Parkinson’s disease. npj Parkinson’s Dis. 2022;8:1–18. - PMC - PubMed
-
- Bates G. Huntingtin aggregation and toxicity in Huntington’s disease. Lancet. 2003;361:1642–1644. - PubMed
MeSH terms
Substances
Grants and funding
- 866004/EC | European Research Council (ERC)
- 823839/European Proteomics Infrastructure Consortium providing access (EPIC-XS)
- 184951/SNSF_/Swiss National Science Foundation/Switzerland
- CRSII5_177195/SNSF_/Swiss National Science Foundation/Switzerland
- MC_U105178783/MRC_/Medical Research Council/United Kingdom
LinkOut - more resources
Full Text Sources
