

A Perspective on Protein Structure Prediction Using Quantum Computers
- PMID: 38703105
- PMCID: PMC11099973
- DOI: 10.1021/acs.jctc.4c00067
A Perspective on Protein Structure Prediction Using Quantum Computers
Abstract
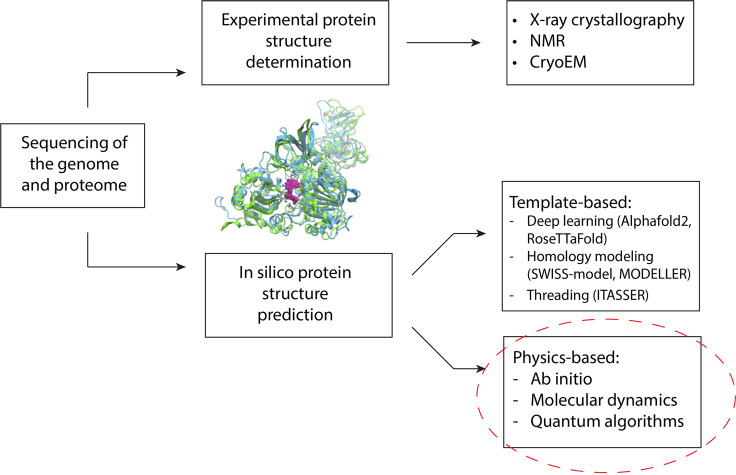
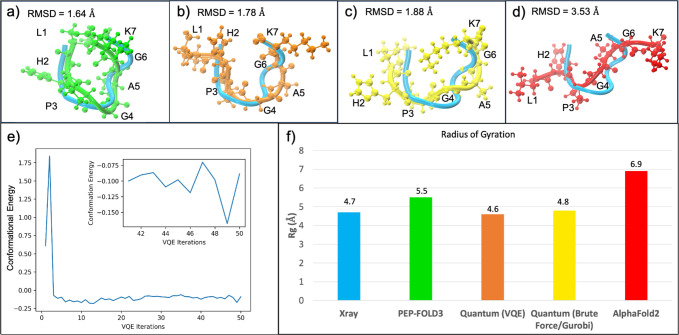
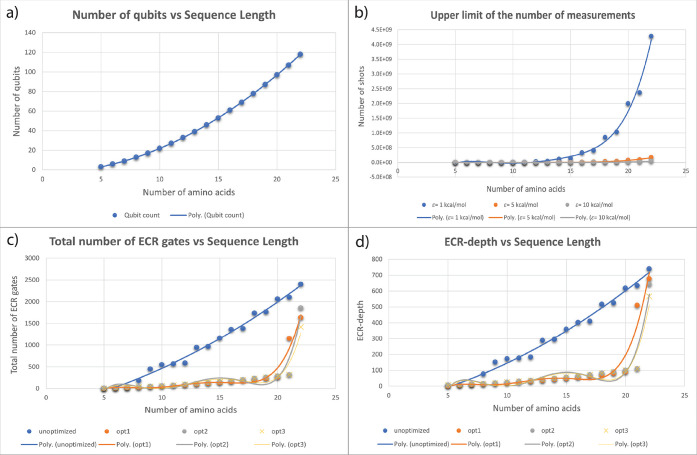
Despite the recent advancements by deep learning methods such as AlphaFold2, in silico protein structure prediction remains a challenging problem in biomedical research. With the rapid evolution of quantum computing, it is natural to ask whether quantum computers can offer some meaningful benefits for approaching this problem. Yet, identifying specific problem instances amenable to quantum advantage and estimating the quantum resources required are equally challenging tasks. Here, we share our perspective on how to create a framework for systematically selecting protein structure prediction problems that are amenable for quantum advantage, and estimate quantum resources for such problems on a utility-scale quantum computer. As a proof-of-concept, we validate our problem selection framework by accurately predicting the structure of a catalytic loop of the Zika Virus NS3 Helicase, on quantum hardware.
Conflict of interest statement
The authors declare no competing financial interest.
Figures
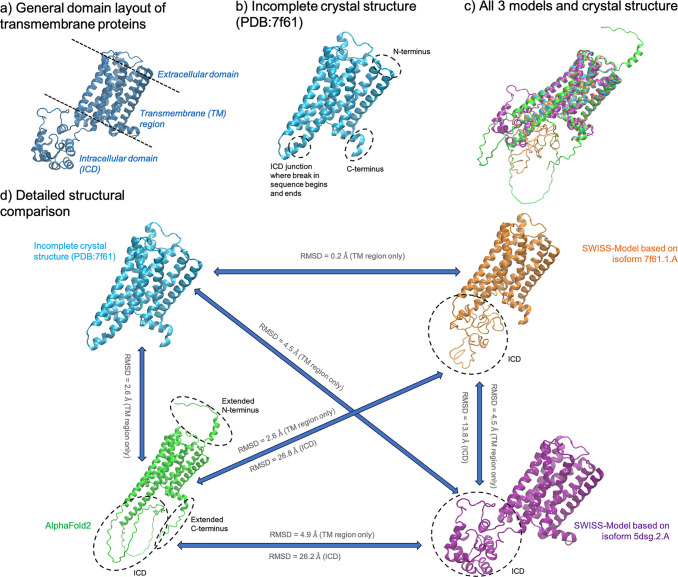

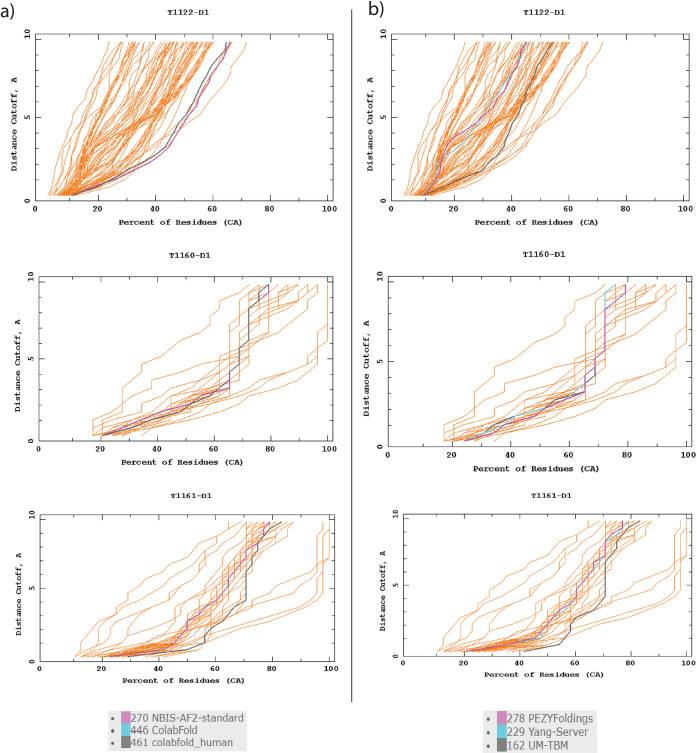
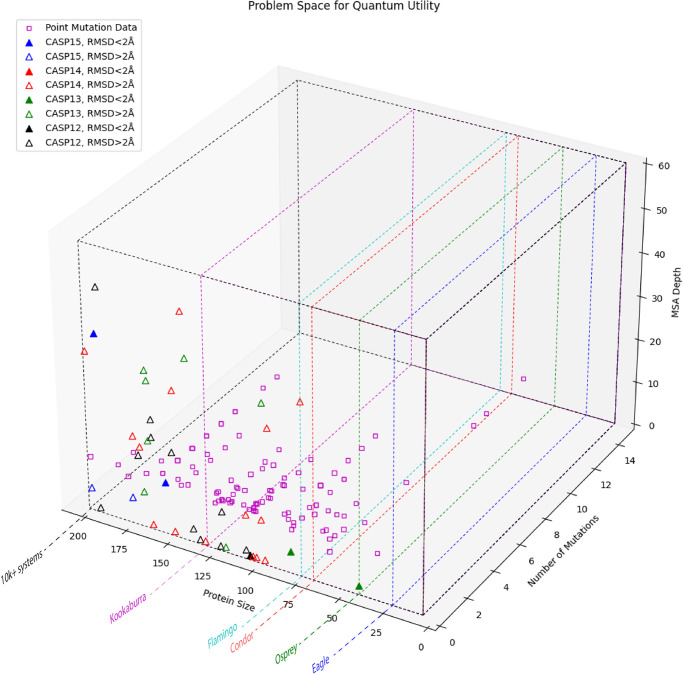
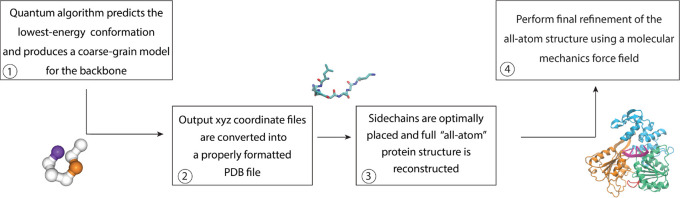
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials