

Assessing personalized responses to anti-PD-1 treatment using patient-derived lung tumor-on-chip
- PMID: 38703767
- PMCID: PMC11148770
- DOI: 10.1016/j.xcrm.2024.101549
Assessing personalized responses to anti-PD-1 treatment using patient-derived lung tumor-on-chip
Abstract
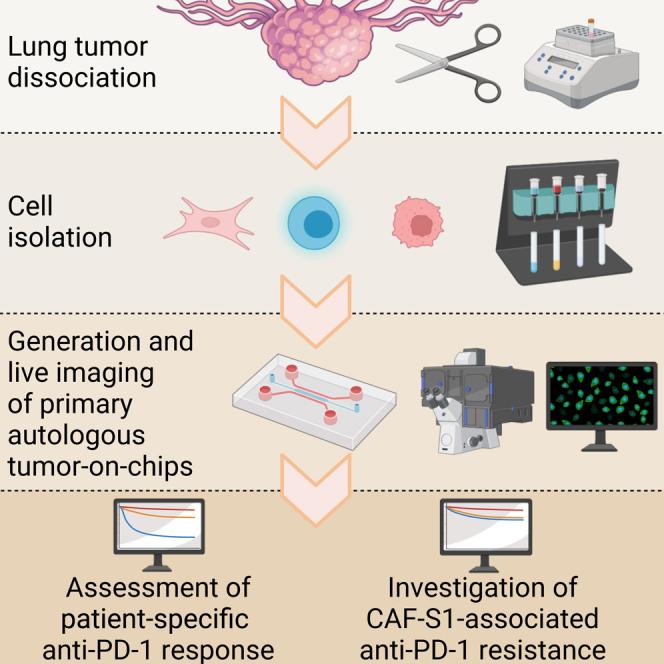
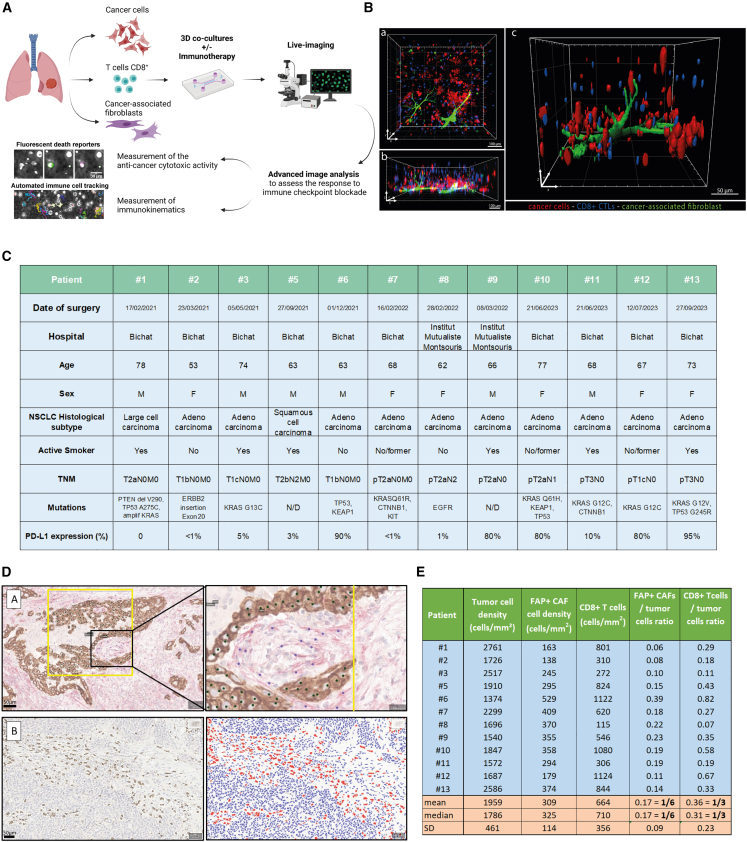
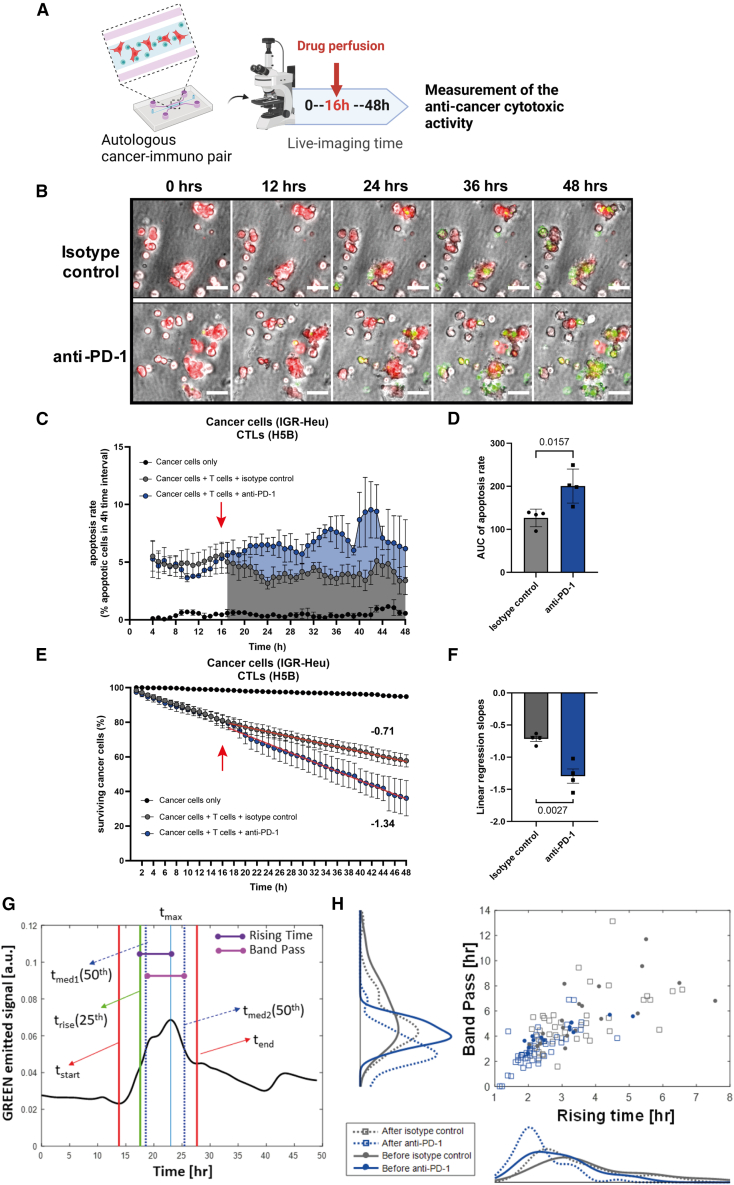
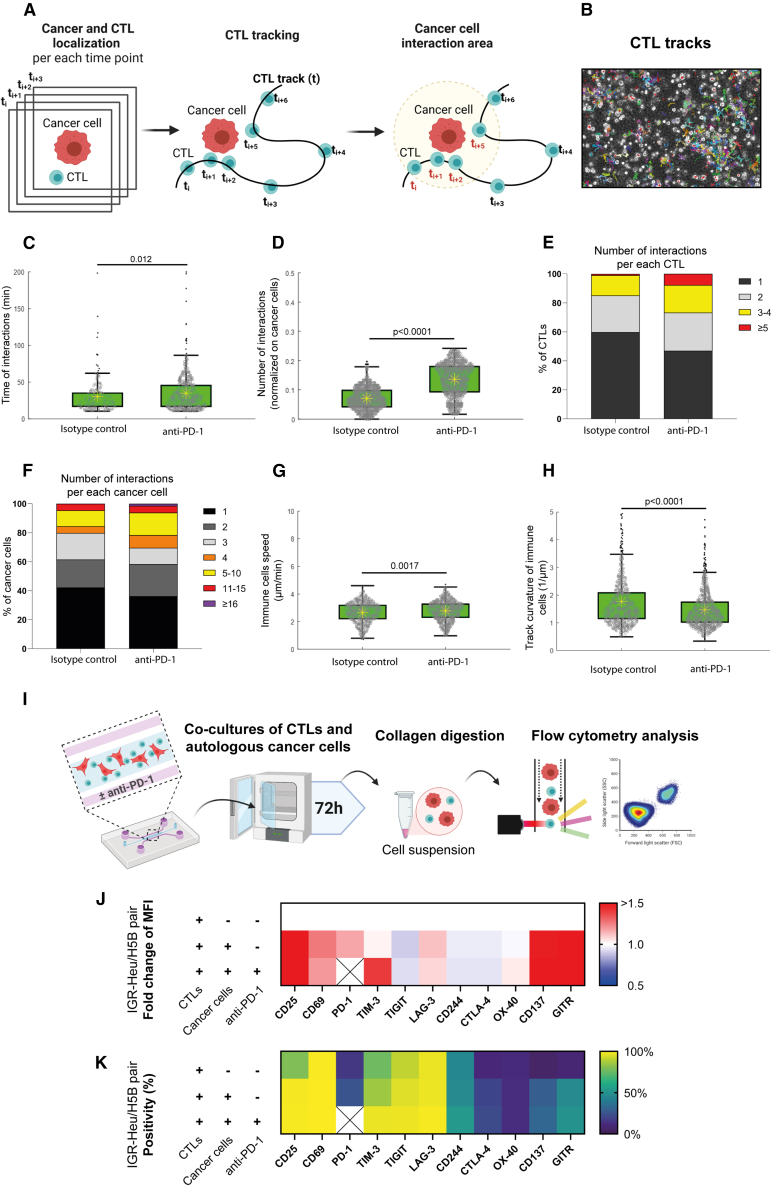
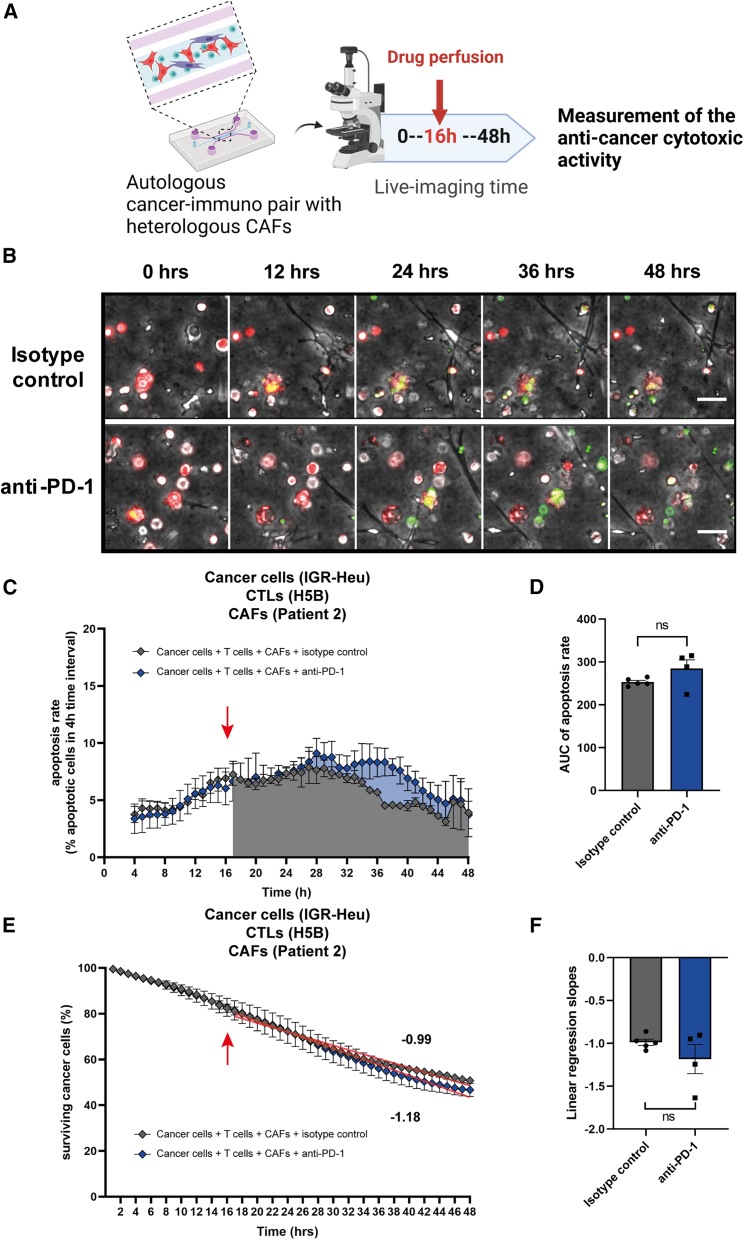
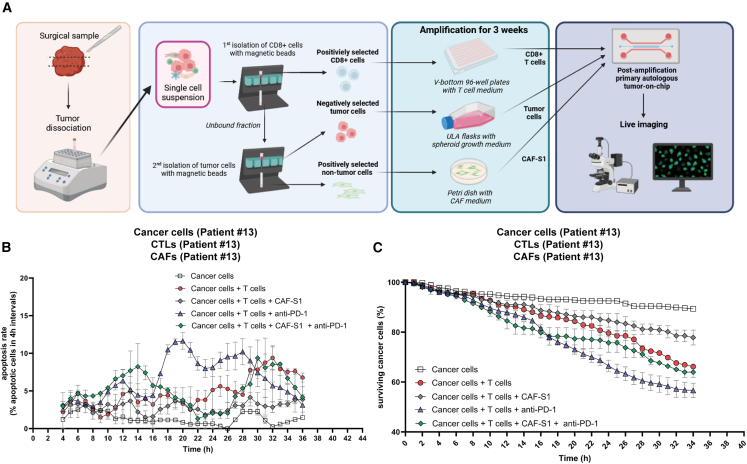
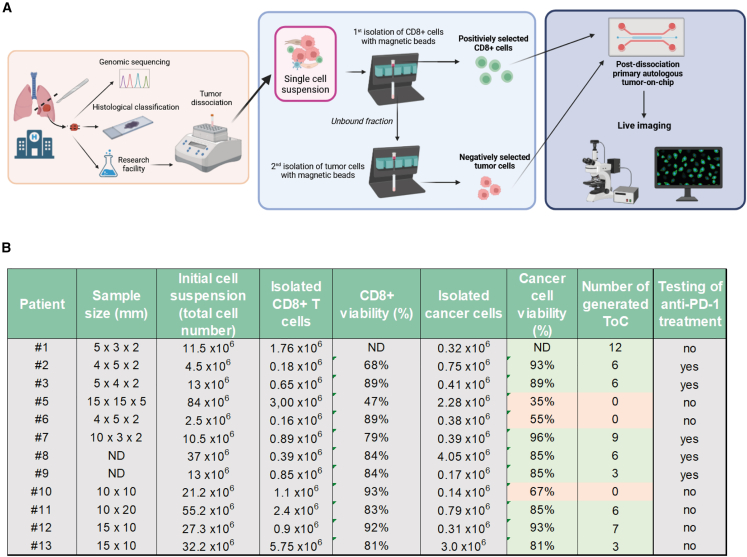
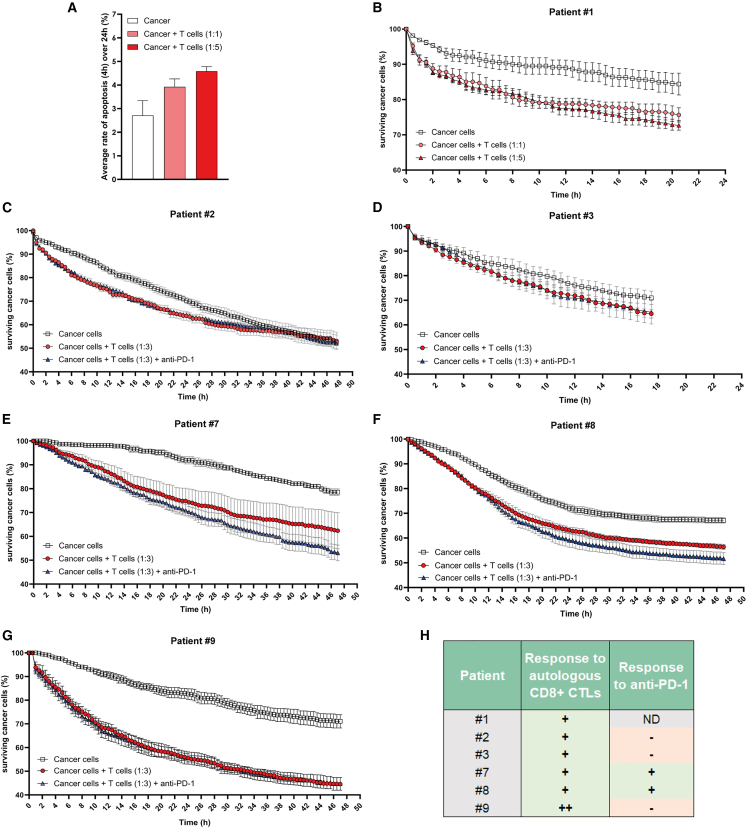
There is a compelling need for approaches to predict the efficacy of immunotherapy drugs. Tumor-on-chip technology exploits microfluidics to generate 3D cell co-cultures embedded in hydrogels that recapitulate simplified tumor ecosystems. Here, we present the development and validation of lung tumor-on-chip platforms to quickly and precisely measure ex vivo the effects of immune checkpoint inhibitors on T cell-mediated cancer cell death by exploiting the power of live imaging and advanced image analysis algorithms. The integration of autologous immunosuppressive FAP+ cancer-associated fibroblasts impaired the response to anti-PD-1, indicating that tumors-on-chips are capable of recapitulating stroma-dependent mechanisms of immunotherapy resistance. For a small cohort of non-small cell lung cancer patients, we generated personalized tumors-on-chips with their autologous primary cells isolated from fresh tumor samples, and we measured the responses to anti-PD-1 treatment. These results support the power of tumor-on-chip technology in immuno-oncology research and open a path to future clinical validations.
Keywords: anti-PD-1; cancer models; cancer-associated fibroblasts; immuno-oncology; immunotherapy; lung cancer; microfluidics; patient-derived; tumor microenvironment; tumor-on-chip.
Copyright © 2024 The Authors. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests H.S. and I.V. are Roche employees. The STAMP method used in this work has been patented by I.V., A.M., E.M., and M.C.P.
Figures
References
-
- Bouquerel C., Dubrova A., Hofer I., Phan D.T.T., Bernheim M., Ladaigue S., Cavaniol C., Maddalo D., Cabel L., Mechta-Grigoriou F., et al. Bridging the gap between tumor-on-chip and clinics: a systematic review of 15 years of studies. Lab Chip. 2023;23:3906–3935. doi: 10.1039/d3lc00531c. - DOI - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
