

A case study of a liver transplant-treated patient with glycogen storage disease type Ia presenting with multiple inflammatory hepatic adenomas: an analysis of clinicopathologic and genetic data
- PMID: 38711024
- PMCID: PMC11075316
- DOI: 10.1186/s12920-024-01888-6
A case study of a liver transplant-treated patient with glycogen storage disease type Ia presenting with multiple inflammatory hepatic adenomas: an analysis of clinicopathologic and genetic data
Abstract
Background: Glycogen storage disease (GSD) is a disease caused by excessive deposition of glycogen in tissues due to genetic disorders in glycogen metabolism. Glycogen storage disease type I (GSD-I) is also known as VonGeirk disease and glucose-6-phosphatase deficiency. This disease is inherited in an autosomal recessive manner, and both sexes can be affected. The main symptoms include hypoglycaemia, hepatomegaly, acidosis, hyperlipidaemia, hyperuricaemia, hyperlactataemia, coagulopathy and developmental delay.
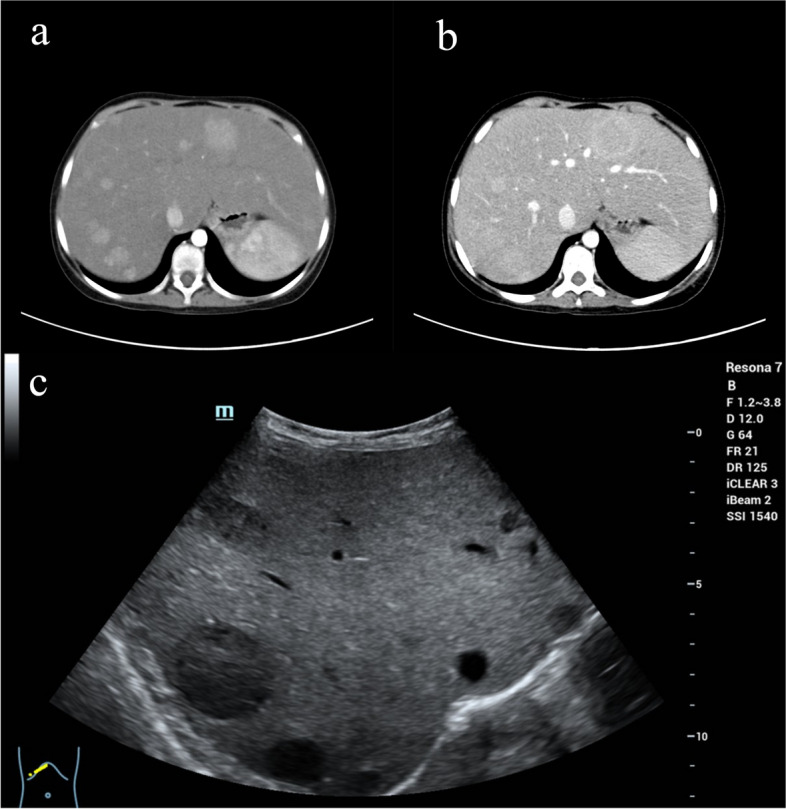
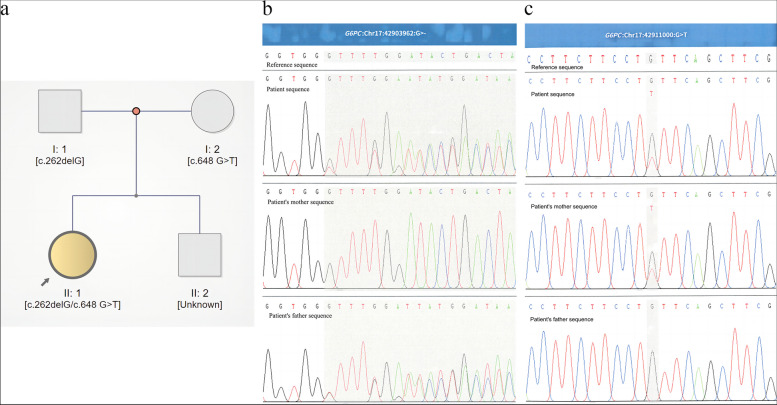
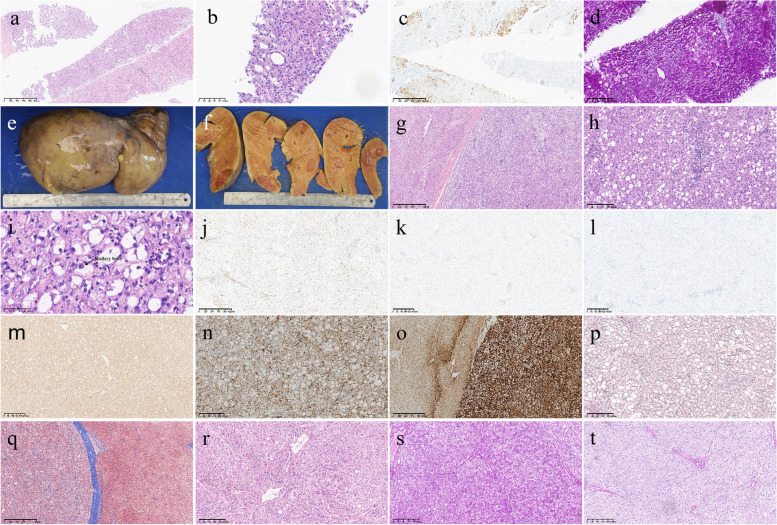
Case presentation: Here, we present the case of a 13-year-old female patient with GSD Ia complicated with multiple inflammatory hepatic adenomas. She presented to the hospital with hepatomegaly, hypoglycaemia, and epistaxis. By clinical manifestations and imaging and laboratory examinations, we suspected that the patient suffered from GSD I. Finally, the diagnosis was confirmed by liver pathology and whole-exome sequencing (WES). WES revealed a synonymous mutation, c.648 G > T (p.L216 = , NM_000151.4), in exon 5 and a frameshift mutation, c.262delG (p.Val88Phefs*14, NM_000151.4), in exon 2 of the G6PC gene. According to the pedigree analysis results of first-generation sequencing, heterozygous mutations of c.648 G > T and c.262delG were obtained from the patient's father and mother. Liver pathology revealed that the solid nodules were hepatocellular hyperplastic lesions, and immunohistochemical (IHC) results revealed positive expression of CD34 (incomplete vascularization), liver fatty acid binding protein (L-FABP) and C-reactive protein (CRP) in nodule hepatocytes and negative expression of β-catenin and glutamine synthetase (GS). These findings suggest multiple inflammatory hepatocellular adenomas. PAS-stained peripheral hepatocytes that were mostly digested by PAS-D were strongly positive. This patient was finally diagnosed with GSD-Ia complicated with multiple inflammatory hepatic adenomas, briefly treated with nutritional therapy after diagnosis and then underwent living-donor liver allotransplantation. After 14 months of follow-up, the patient recovered well, liver function and blood glucose levels remained normal, and no complications occurred.
Conclusion: The patient was diagnosed with GSD-Ia combined with multiple inflammatory hepatic adenomas and received liver transplant treatment. For childhood patients who present with hepatomegaly, growth retardation, and laboratory test abnormalities, including hypoglycaemia, hyperuricaemia, and hyperlipidaemia, a diagnosis of GSD should be considered. Gene sequencing and liver pathology play important roles in the diagnosis and typing of GSD.
Keywords: Clinicopathologic; G6PC gene; GSD-Ia; Hepatic adenomas; Liver transplant-treated.
© 2024. The Author(s).
Conflict of interest statement
Competing interests. The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Grants and funding
- PYYZ-2022-03/Panzhihua City Medical Research Center Project
- PYYZ-2022-03/Panzhihua City Medical Research Center Project
- PYYZ-2022-03/Panzhihua City Medical Research Center Project
- PYYZ-2022-03/Panzhihua City Medical Research Center Project
- PYYZ-2022-03/Panzhihua City Medical Research Center Project
- 2021YJ0168/Sichuan Province Science and Technology Support Program
- 2021YJ0168/Sichuan Province Science and Technology Support Program
- 2021YJ0168/Sichuan Province Science and Technology Support Program
- 2021YJ0168/Sichuan Province Science and Technology Support Program
- 2021YJ0168/Sichuan Province Science and Technology Support Program
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
