

On the Validation of Protein Force Fields Based on Structural Criteria
- PMID: 38711373
- PMCID: PMC11103706
- DOI: 10.1021/acs.jpcb.3c08469
On the Validation of Protein Force Fields Based on Structural Criteria
Abstract
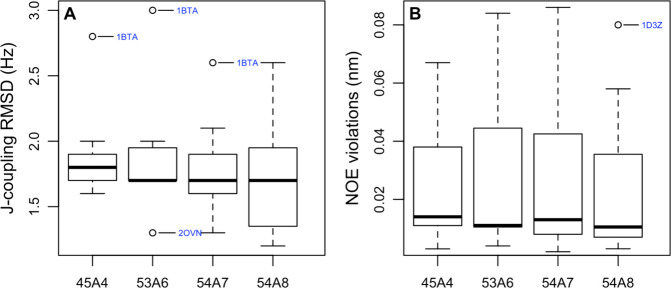
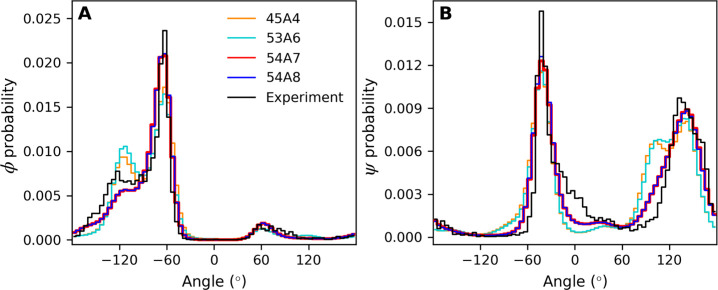
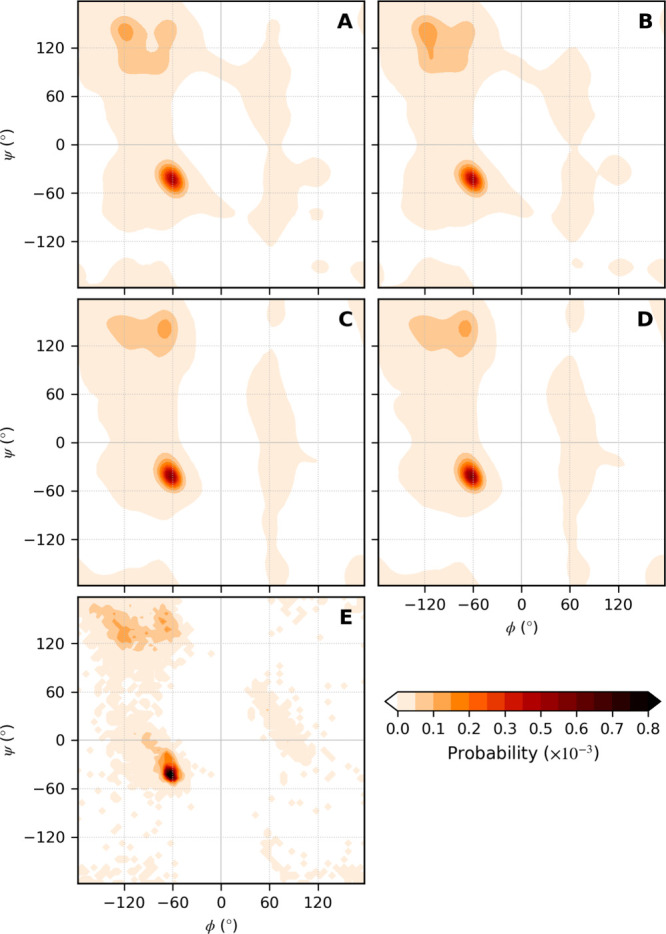
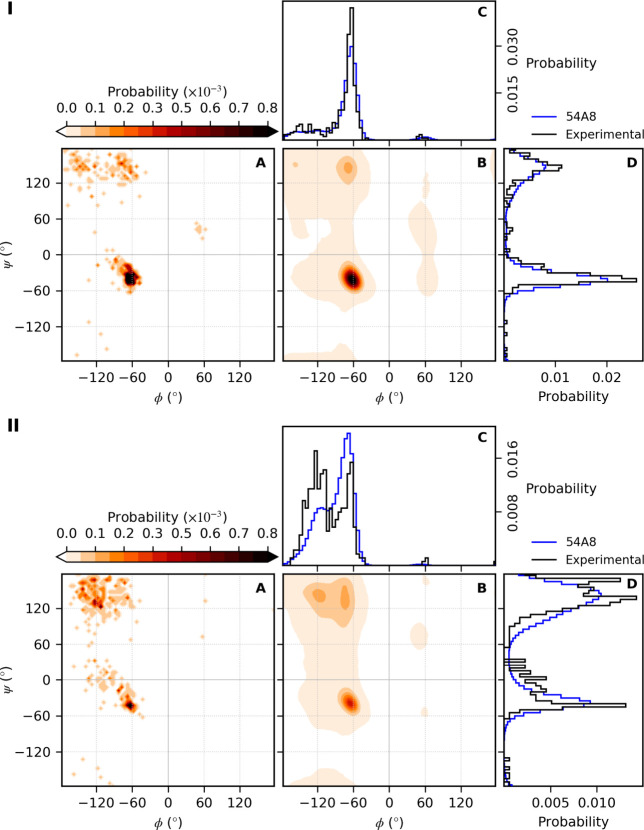
Molecular dynamics simulations depend critically on the quality of the force field used to describe the interatomic interactions and the extent to which it has been validated for use in a specific application. Using a curated test set of 52 high-resolution structures, 39 derived from X-ray diffraction and 13 solved using NMR, we consider the extent to which different parameter sets of the GROMOS protein force field can be distinguished based on comparing a range of structural criteria, including the number of backbone hydrogen bonds, the number of native hydrogen bonds, polar and nonpolar solvent-accessible surface area, radius of gyration, the prevalence of secondary structure elements, J-coupling constants, nuclear Overhauser effect (NOE) intensities, positional root-mean-square deviations (RMSD), and the distribution of backbone ϕ and ψ dihedral angles. It is shown that while statistically significant differences between the average values of individual metrics could be detected, these were in general small. Furthermore, improvements in agreement in one metric were often offset by loss of agreement in another. The work establishes a framework and test set against which protein force fields can be validated. It also highlights the danger of inferring the relative quality of a given force field based on a small range of structural properties or small number of proteins.
Conflict of interest statement
The authors declare no competing financial interest.
Figures

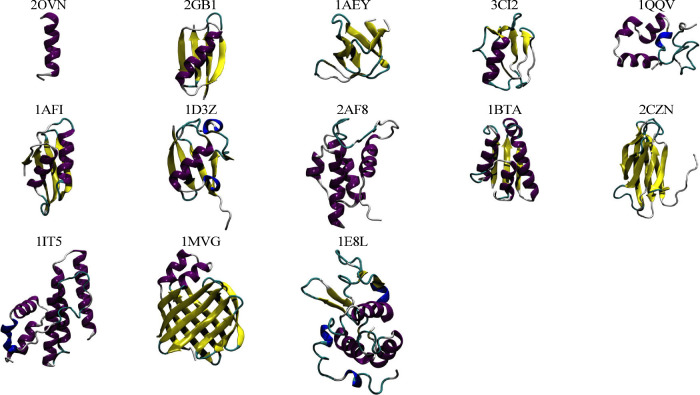
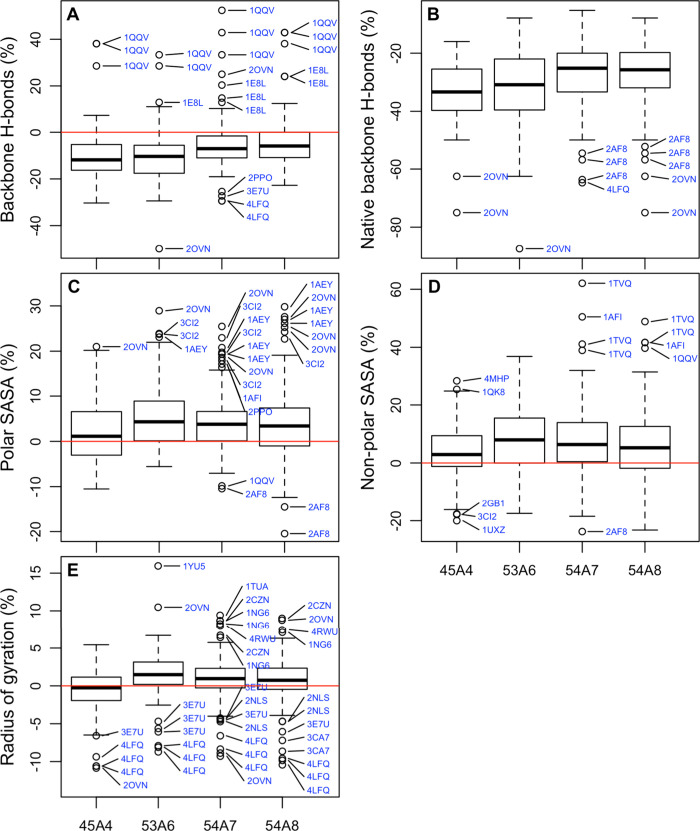
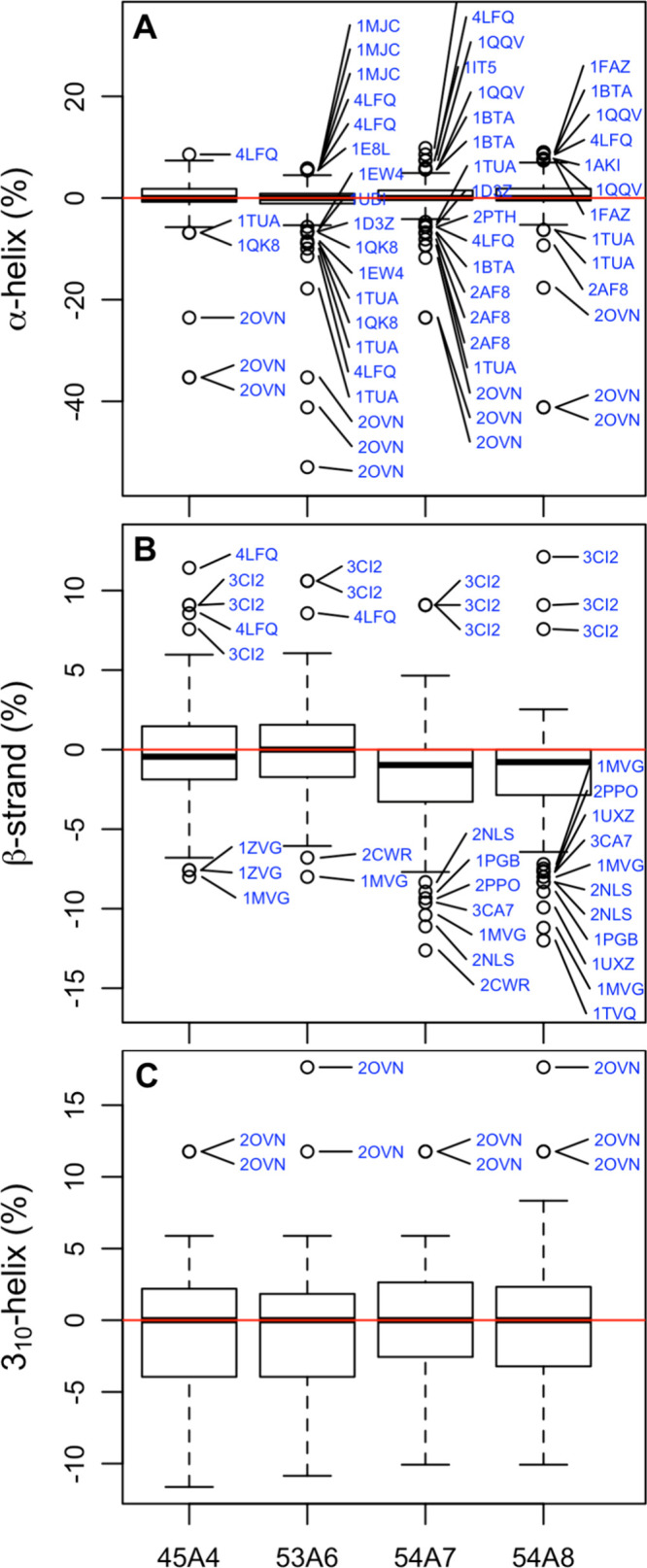
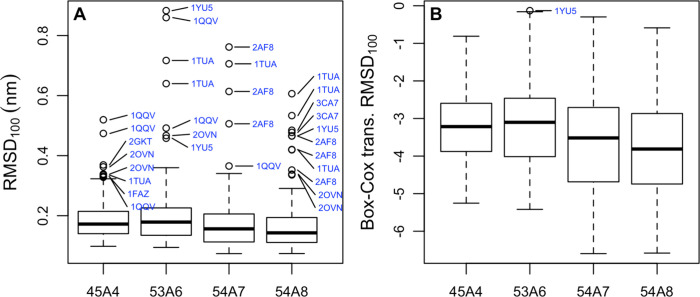
References
-
- MacKerell A. D. Jr.; Bashford D.; Bellott M.; Dunbrack R. L. Jr.; Evanseck J. D.; Field M. J.; Fischer S.; Gao J.; Guo H.; Ha S.; et al. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B 1998, 102 (18), 3586–3616. 10.1021/jp973084f. - DOI - PubMed
-
- Cornell W. D.; Cieplak P.; Bayly C. I.; Gould I. R.; Merz K. M.; Ferguson D. M.; Spellmeyer D. C.; Fox T.; Caldwell J. W.; Kollman P. A. A second generation force field for the simulation of proteins, nucleic acids, and organic molecules. J. Am. Chem. Soc. 1995, 117 (19), 5179–5197. 10.1021/ja00124a002. - DOI
-
- Jorgensen W. L.; Maxwell D. S.; Tirado-Rives J. Development and testing of the OPLS all-atom force field on conformational energetics and properties of organic liquids. J. Am. Chem. Soc. 1996, 118 (45), 11225–11236. 10.1021/ja9621760. - DOI
-
- Scott W. R. P.; Hunenberger P. H.; Tironi I. G.; Mark A. E.; Billeter S. R.; Fennen J.; Torda A. E.; Huber T.; Kruger P.; van Gunsteren W. F. The GROMOS biomolecular simulation program package. J. Phys. Chem. A 1999, 103 (19), 3596–3607. 10.1021/jp984217f. - DOI
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
