

FGFR inhibition blocks NF-ĸB-dependent glucose metabolism and confers metabolic vulnerabilities in cholangiocarcinoma
- PMID: 38714664
- PMCID: PMC11076599
- DOI: 10.1038/s41467-024-47514-y
FGFR inhibition blocks NF-ĸB-dependent glucose metabolism and confers metabolic vulnerabilities in cholangiocarcinoma
Abstract
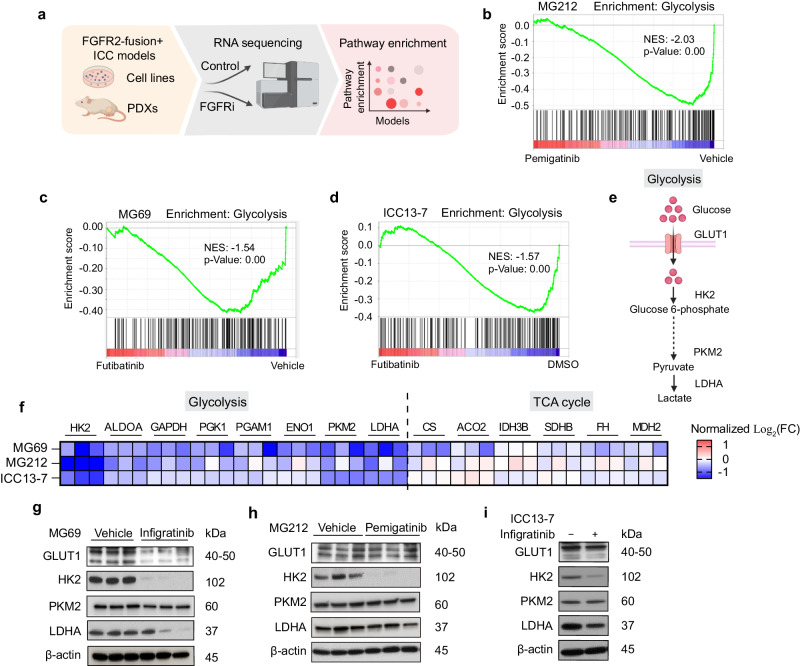
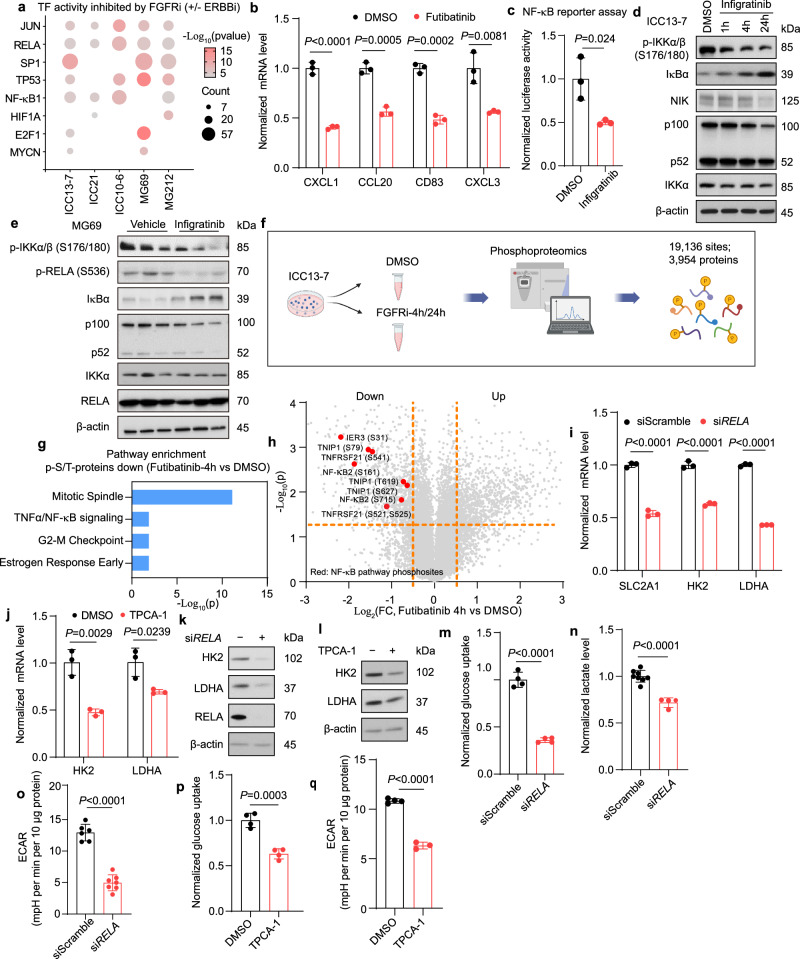
Genomic alterations that activate Fibroblast Growth Factor Receptor 2 (FGFR2) are common in intrahepatic cholangiocarcinoma (ICC) and confer sensitivity to FGFR inhibition. However, the depth and duration of response is often limited. Here, we conduct integrative transcriptomics, metabolomics, and phosphoproteomics analysis of patient-derived models to define pathways downstream of oncogenic FGFR2 signaling that fuel ICC growth and to uncover compensatory mechanisms associated with pathway inhibition. We find that FGFR2-mediated activation of Nuclear factor-κB (NF-κB) maintains a highly glycolytic phenotype. Conversely, FGFR inhibition blocks glucose uptake and glycolysis while inciting adaptive changes, including switching fuel source utilization favoring fatty acid oxidation and increasing mitochondrial fusion and autophagy. Accordingly, FGFR inhibitor efficacy is potentiated by combined mitochondrial targeting, an effect enhanced in xenograft models by intermittent fasting. Thus, we show that oncogenic FGFR2 signaling drives NF-κB-dependent glycolysis in ICC and that metabolic reprogramming in response to FGFR inhibition confers new targetable vulnerabilities.
© 2024. The Author(s).
Conflict of interest statement
N.B. reports research grant support from Kinnate Biopharma, Tyra Biosciences, and Servier Laboratories. D.J. reports grants and personal fees from Novartis, Genentech, Syros, and Eisai, grants from Pfizer, Ribon Therapeutics, Infinity, InventisBio, Cyteir, and Arvinas, and personal fees from Relay Therapeutics, Vibliome, Mapkure, and PIC Therapeutics outside the submitted work. The remaining authors declare no competing interests.
Figures



References
-
- Oh, D.-Y. et al. Durvalumab plus Gemcitabine and Cisplatin in advanced biliary tract cancer. NEJM Evid.10.1056/EVIDoa2200015 (2022). - PubMed
-
- Cleary J. Extracellular domain in-frame deletions are therapeutically targetable genomic alterations that function as oncogenic drivers in cholangiocarcinoma. Cancer Discov. 2021;11:2488–2505. doi: 10.1158/2159-8290.CD-20-1669. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
