

Longitudinal molecular profiling elucidates immunometabolism dynamics in breast cancer
- PMID: 38714665
- PMCID: PMC11076527
- DOI: 10.1038/s41467-024-47932-y
Longitudinal molecular profiling elucidates immunometabolism dynamics in breast cancer
Abstract
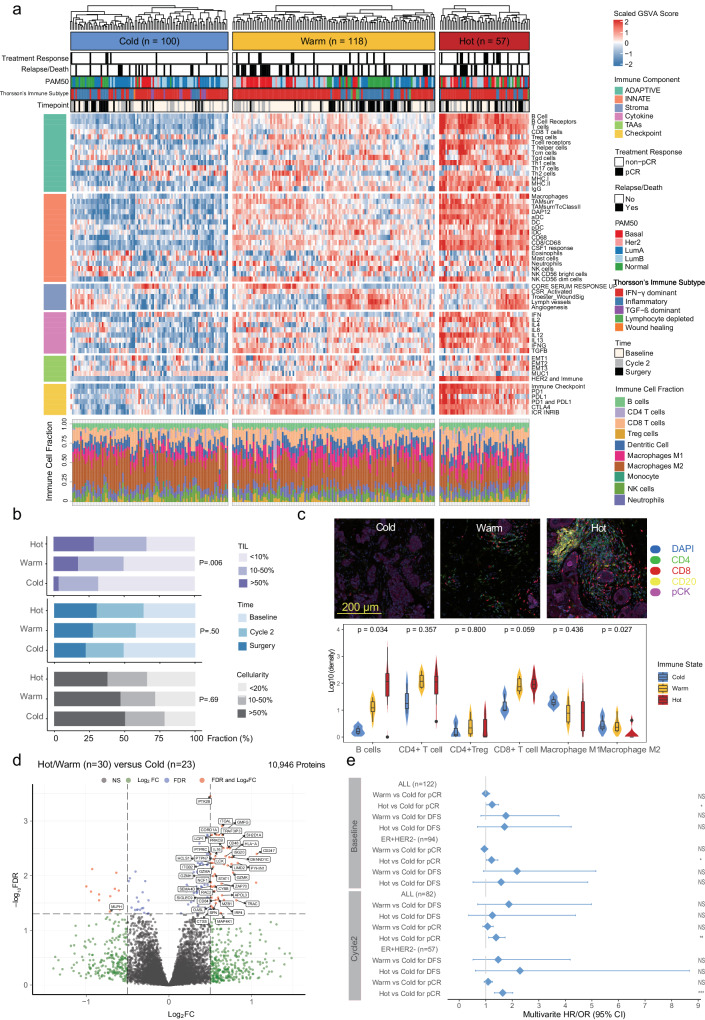
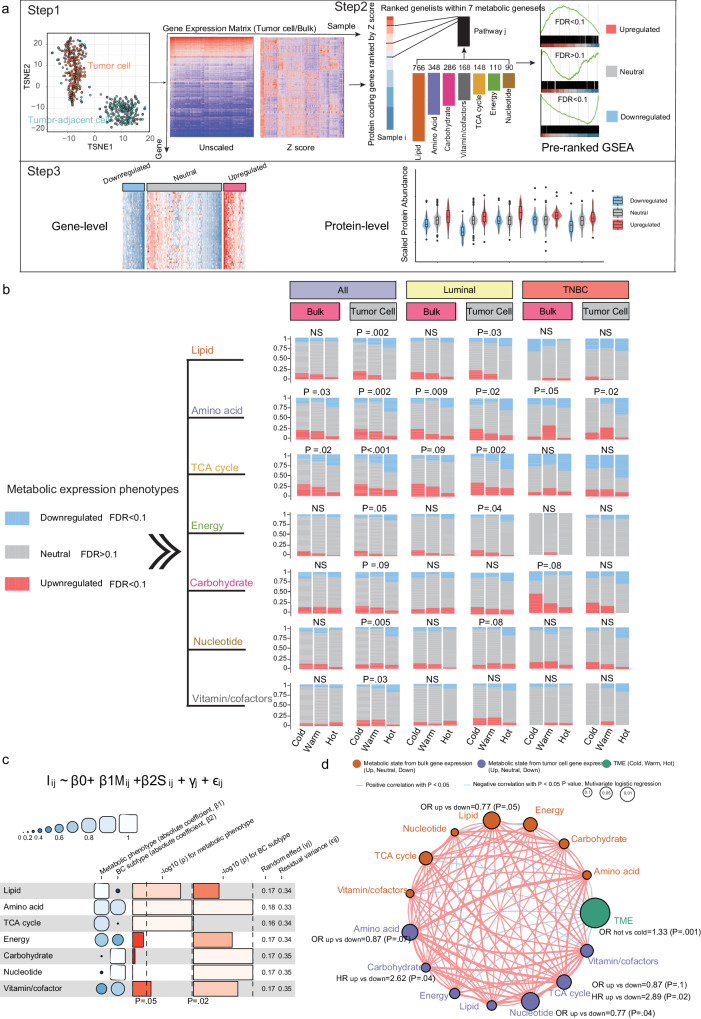
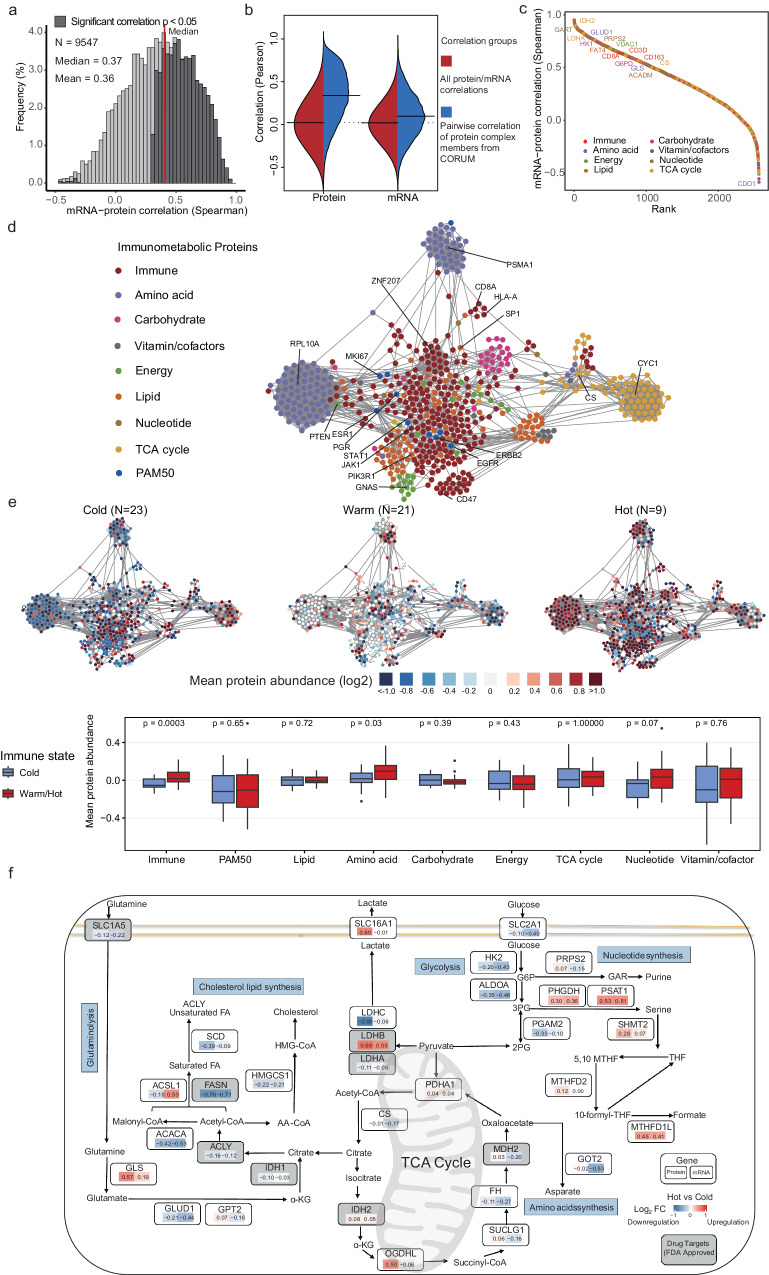
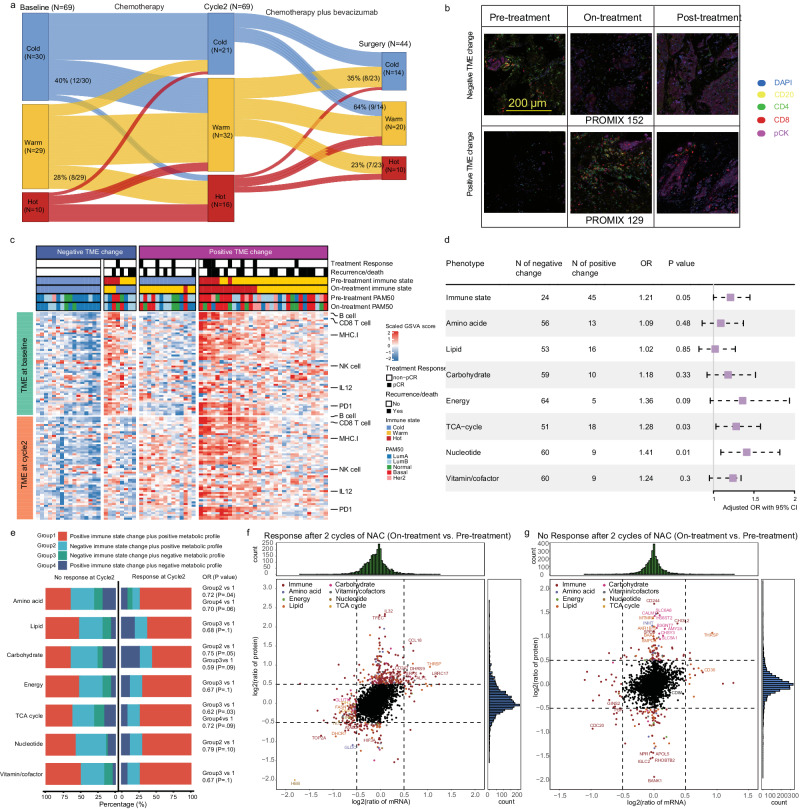
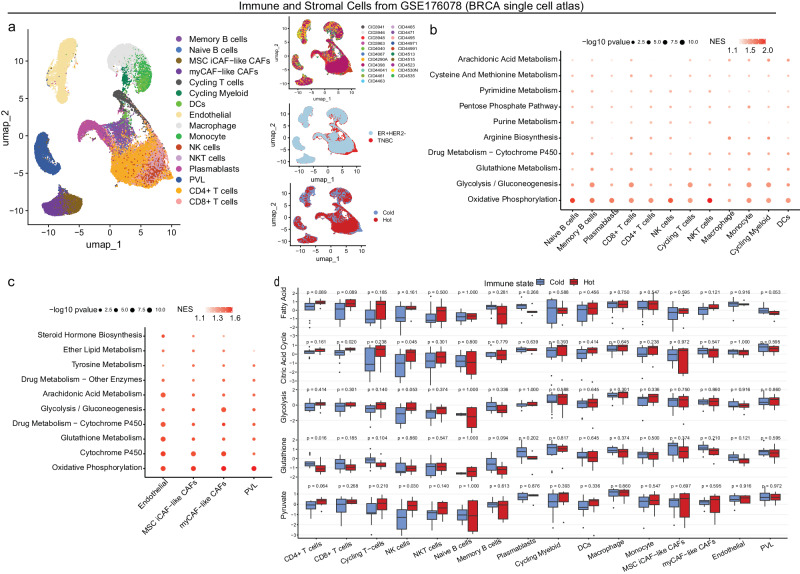
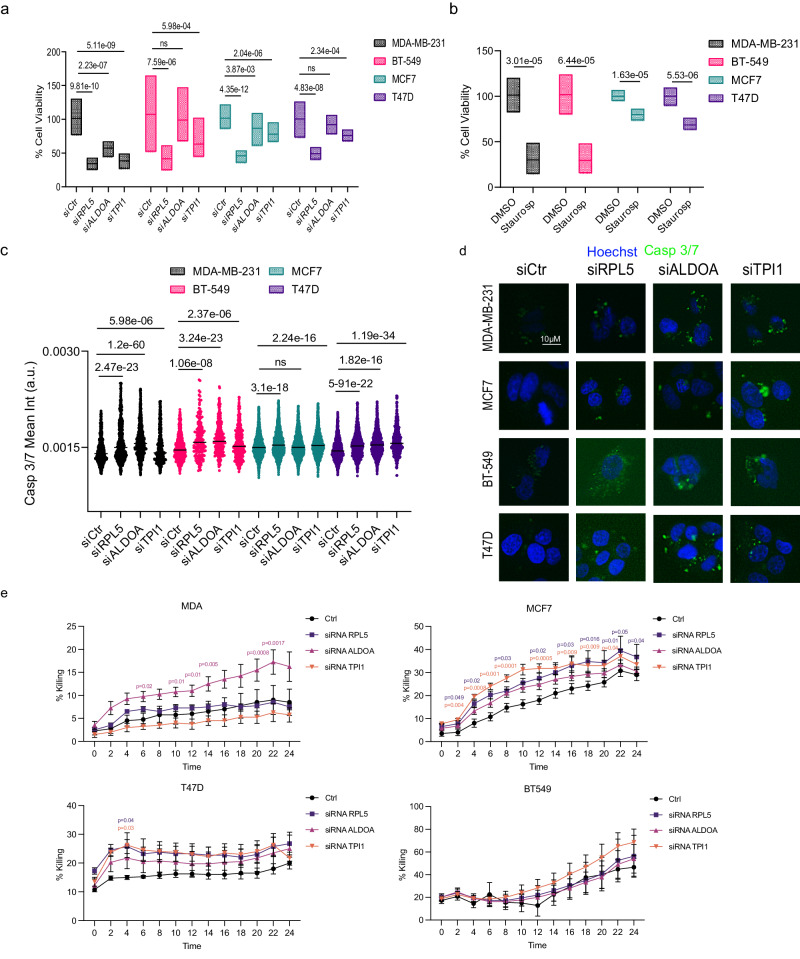
Although metabolic reprogramming within tumor cells and tumor microenvironment (TME) is well described in breast cancer, little is known about how the interplay of immune state and cancer metabolism evolves during treatment. Here, we characterize the immunometabolic profiles of tumor tissue samples longitudinally collected from individuals with breast cancer before, during and after neoadjuvant chemotherapy (NAC) using proteomics, genomics and histopathology. We show that the pre-, on-treatment and dynamic changes of the immune state, tumor metabolic proteins and tumor cell gene expression profiling-based metabolic phenotype are associated with treatment response. Single-cell/nucleus RNA sequencing revealed distinct tumor and immune cell states in metabolism between cold and hot tumors. Potential drivers of NAC based on above analyses were validated in vitro. In summary, the study shows that the interaction of tumor-intrinsic metabolic states and TME is associated with treatment outcome, supporting the concept of targeting tumor metabolism for immunoregulation.
© 2024. The Author(s).
Conflict of interest statement
Jonas Bergh reports grants from Amgen, AstraZeneca, Bayer, Merck, Pfizer, Roche, and Sanofi-Aventis outside the submitted work, as well as honoraria to Asklepios Medicine HB. In addition, Jonas Bergh is co-author on a chapter on prognostic and predictive factors in early, non-metastatic breast cancer in UpToDate; Jonas Bergh has also been offered stocks in Stratipath and offered to be consultant for that diagnostic company in early development. Niklas Loman reports honoraria for educational proposes and advisory boards received by his employer from Astra Zeneca, MSD, Roche, Gilead, and MSD; Janne Lehtio reports other support from Fenomark Diagnostics outside the submitted work; Janne Lehtio is involved in Cancer Core Europe BoB trial financed by Roche (not related to this work). Alexios Matikas reports other support from Veracyte and Roche outside the submitted work. Theodoros Foukakis reports institutional grants and personal fees from Novartis; institutional grants and personal fees from Pfizer; institutional grants from AstraZeneca; personal fees from Affibody, Exact Sciences, UpToDate and Veracyte; honoraria paid to his institution from Gilead, Pfizer, AstraZeneca and Roche outside the submitted work. No disclosures were reported by the other authors.
Figures



References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Medical
