

Determinants of mosaic chromosomal alteration fitness
- PMID: 38714703
- PMCID: PMC11076528
- DOI: 10.1038/s41467-024-48190-8
Determinants of mosaic chromosomal alteration fitness
Abstract
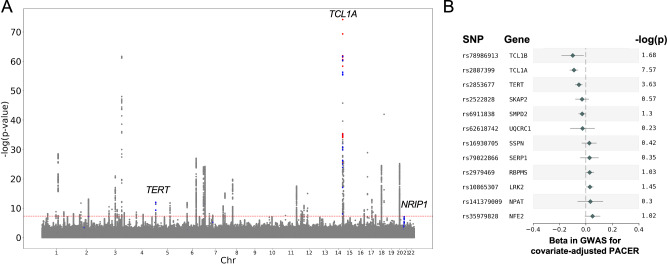
Clonal hematopoiesis (CH) is characterized by the acquisition of a somatic mutation in a hematopoietic stem cell that results in a clonal expansion. These driver mutations can be single nucleotide variants in cancer driver genes or larger structural rearrangements called mosaic chromosomal alterations (mCAs). The factors that influence the variations in mCA fitness and ultimately result in different clonal expansion rates are not well understood. We used the Passenger-Approximated Clonal Expansion Rate (PACER) method to estimate clonal expansion rate as PACER scores for 6,381 individuals in the NHLBI TOPMed cohort with gain, loss, and copy-neutral loss of heterozygosity mCAs. Our mCA fitness estimates, derived by aggregating per-individual PACER scores, were correlated (R2 = 0.49) with an alternative approach that estimated fitness of mCAs in the UK Biobank using population-level distributions of clonal fraction. Among individuals with JAK2 V617F clonal hematopoiesis of indeterminate potential or mCAs affecting the JAK2 gene on chromosome 9, PACER score was strongly correlated with erythrocyte count. In a cross-sectional analysis, genome-wide association study of estimates of mCA expansion rate identified a TCL1A locus variant associated with mCA clonal expansion rate, with suggestive variants in NRIP1 and TERT.
© 2024. The Author(s).
Conflict of interest statement
A.G.B. and S.J. are cofounders, equity holders, and on the scientific advisory board of TenSixteen Bio. M.H.C. has received grant support from Bayer. B.M.P. serves on the Steering Committee of the Yale Open Data Access Project funded by Johnson & Johnson. Stanford University has filed a patent application for the use of PACER to identify therapeutic targets on which S.J., A.G.B. and J.S.W. are inventors (US patent 63/141,333). The patent has been licensed to TenSixteen Bio. L.M.R. is a consultant for the NHLBI TOPMed Administrative Coordinating Center (through Westat). S.S. Rich is a consultant to Westat for NHLBI TOPMed. In the past three years, E.K.S. received grant support from Bayer and Northpond Laboratories. All other authors declare that they have no competing interests.
Figures




Update of
-
Determinants of mosaic chromosomal alteration fitness.medRxiv [Preprint]. 2023 Oct 21:2023.10.20.23297280. doi: 10.1101/2023.10.20.23297280. medRxiv. 2023. Update in: Nat Commun. 2024 May 7;15(1):3800. doi: 10.1038/s41467-024-48190-8. PMID: 37905118 Free PMC article. Updated. Preprint.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
