

Mapping and functional characterization of structural variation in 1060 pig genomes
- PMID: 38715020
- PMCID: PMC11075355
- DOI: 10.1186/s13059-024-03253-3
Mapping and functional characterization of structural variation in 1060 pig genomes
Abstract
Background: Structural variations (SVs) have significant impacts on complex phenotypes by rearranging large amounts of DNA sequence.
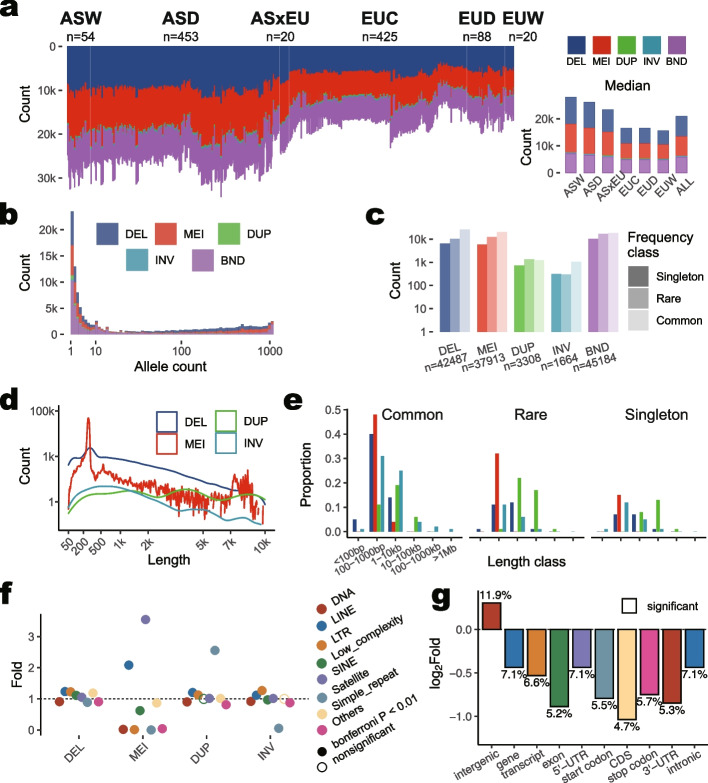
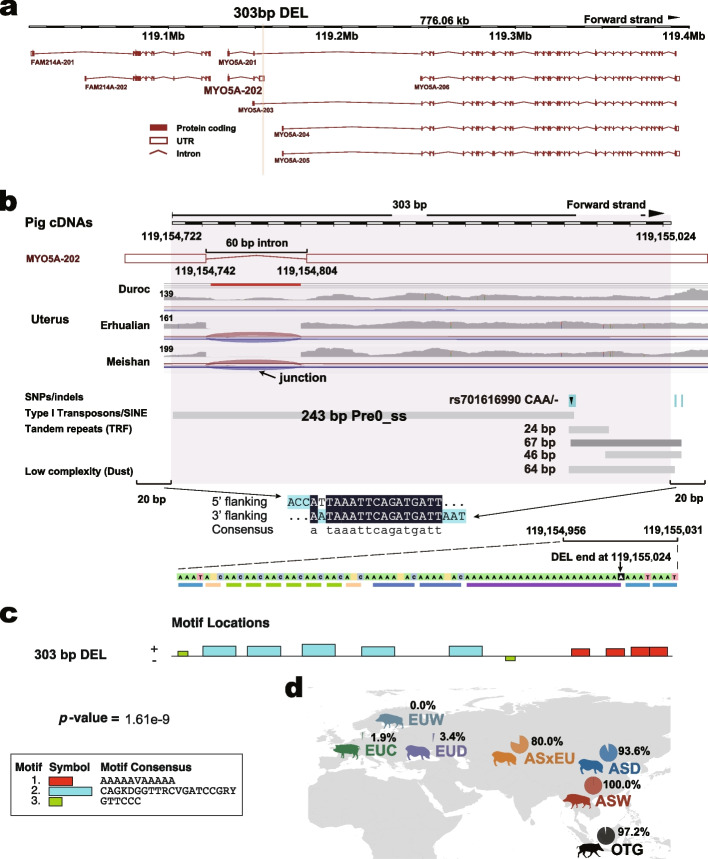
Results: We present a comprehensive SV catalog based on the whole-genome sequence of 1060 pigs (Sus scrofa) representing 101 breeds, covering 9.6% of the pig genome. This catalog includes 42,487 deletions, 37,913 mobile element insertions, 3308 duplications, 1664 inversions, and 45,184 break ends. Estimates of breed ancestry and hybridization using genotyped SVs align well with those from single nucleotide polymorphisms. Geographically stratified deletions are observed, along with known duplications of the KIT gene, responsible for white coat color in European pigs. Additionally, we identify a recent SINE element insertion in MYO5A transcripts of European pigs, potentially influencing alternative splicing patterns and coat color alterations. Furthermore, a Yorkshire-specific copy number gain within ABCG2 is found, impacting chromatin interactions and gene expression across multiple tissues over a stretch of genomic region of ~200 kb. Preliminary investigations into SV's impact on gene expression and traits using the Pig Genotype-Tissue Expression (PigGTEx) data reveal SV associations with regulatory variants and gene-trait pairs. For instance, a 51-bp deletion is linked to the lead eQTL of the lipid metabolism regulating gene FADS3, whose expression in embryo may affect loin muscle area, as revealed by our transcriptome-wide association studies.
Conclusions: This SV catalog serves as a valuable resource for studying diversity, evolutionary history, and functional shaping of the pig genome by processes like domestication, trait-based breeding, and adaptive evolution.
Keywords: Functional genome; Gene expression; Pig; Population diversity; Structure variation.
© 2024. This is a U.S. Government work and not under copyright protection in the US; foreign copyright protection may apply.
Conflict of interest statement
All authors declare that they have no competing interests.
Figures





References
-
- Frantz LA, Schraiber JG, Madsen O, Megens HJ, Bosse M, Paudel Y, Semiadi G, Meijaard E, Li N, Crooijmans RP, et al. Genome sequencing reveals fine scale diversification and reticulation history during speciation in Sus. Genome Biol. 2013;14(9):R107. doi: 10.1186/gb-2013-14-9-r107. - DOI - PMC - PubMed
Publication types
MeSH terms
Grants and funding
- 2021YFF1000600/Key Technologies Research and Development Program
- 2019-67015-29321/National Institute of Food and Agriculture
- 2021-67015-33409/National Institute of Food and Agriculture
- 801215/H2020 Marie Skłodowska-Curie Actions
- 32302699/The Young Scientists Fund of the National Natural Science Foundation of China
