

A roadmap to precision treatments for familial pulmonary fibrosis
- PMID: 38718684
- PMCID: PMC11096859
- DOI: 10.1016/j.ebiom.2024.105135
A roadmap to precision treatments for familial pulmonary fibrosis
Abstract
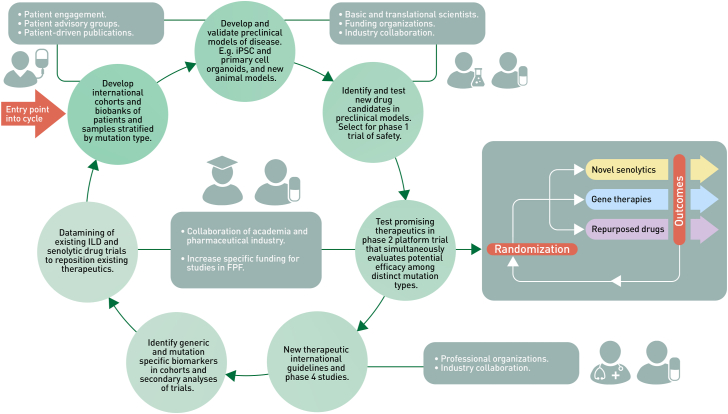
Interstitial lung diseases (ILDs) in adults and children (chILD) are a heterogeneous group of lung disorders leading to inflammation, abnormal tissue repair and scarring of the lung parenchyma often resulting in respiratory failure and death. Inherited factors directly cause, or contribute significantly to the risk of developing ILD, so called familial pulmonary fibrosis (FPF), and monogenic forms may have a poor prognosis and respond poorly to current treatments. Specific, variant-targeted or precision treatments are lacking. Clinical trials of repurposed drugs, anti-fibrotic medications and specific treatments are emerging but for many patients no interventions exist. We convened an expert working group to develop an overarching framework to address the existing research gaps in basic, translational, and clinical research and identified areas for future development of preclinical models, candidate medications and innovative clinical trials. In this Position Paper, we summarise working group discussions, recommendations, and unresolved questions concerning precision treatments for FPF.
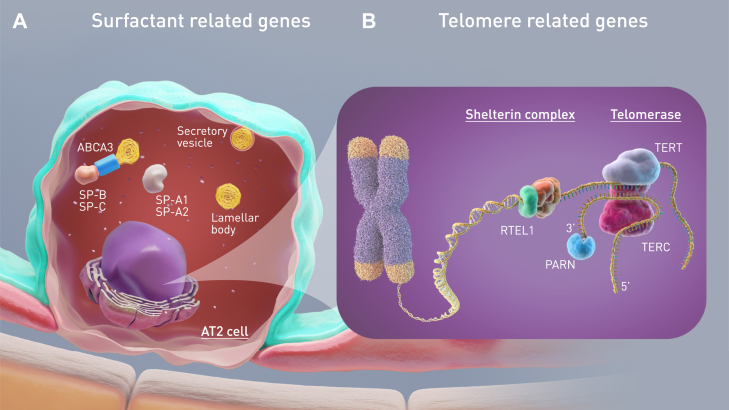
Keywords: Familial pulmonary fibrosis; Induced pluripotent stem cells; Interstitial lung disease of genetic cause, precision medicine; Preclinical models; Pulmonary fibrosis; Surfactant related gene; Telomere related gene; Telomeropathy.
Copyright © 2024 The Author(s). Published by Elsevier B.V. All rights reserved.
Conflict of interest statement
Declaration of interests No authors have stocks or shares, equity, a contract of employment, or a named position on a company board. KH reports grants from COST (European Cooperation in Science and Technology) Innovator Grant (CIG 16125) and the HRB Emerging Clinical Scientist Award (ECSA-2020-011) which supported the manuscript. KH also reports grants from Moderna Tx, and lecturer fees from Boehringer Ingelheim and PatientMPower outside of the submitted work. MO reports grants from the Irish Research Council and support for attending meetings from the Irish Thoracic Society and GlaxoKleinSmith outside of the submitted work. QP reports support for attending meetings from Janssen and lecturer fees from Gilead outside of the submitted work. LG reports a paid leadership role in the EU-IPFF and unrestricted organisational grants to EU-IPFF from Bristol Meyer Squibb, Boehringer Ingelheim, Chiesi Pharmaceuticals, Ferrar Pharmaceuticals, CSL Behring, Trevi Therapeutics, The Roche Group, Vicore Pharma. LG also reports support for attending meetings from the ERS, European Lung Foundation, European Reference Network on Rare Respiratory Diseases and unpaid leadership roles in European Reference Network on Rare Respiratory Diseases, European Lung Foundation, the Irish Lung Fibrosis Association. GJ reports grants from Astra Zeneca, Biogen, Galecto, GlaxoSmithKline, Nordic Biosciences, RedX, Pliant, consulting fees from Astra Zeneca, Brainomix, Bristol Myers Squibb, Chiesi, Cohbar, Daewoong, GlaxoSmithKline, Veracyte, Resolution Therapeutics, Pliant, and lecturer fees from Boehringer Ingelheim, Chiesi, Roche PatientMPower, AstraZeneca outside of the submitted work. GJ also reports payments from Pinsent Masons LLP for expert testimony, participation on advisory board for Boehringer Ingelheim, Galapagos, Vicore, and leadership roles for NuMedii and Action for Pulmonary Fibrosis. MG reports grants, participation on advisory and adjudication boards, and lecturer fees from Boehringer Ingelheim outside of the submitted work. NN reports grants from Million Dollar Bike Ride, Chancellerie des Universités: Legs Poix, (n°2022000594), support for attending meetings from the ERS and an unpaid leadership role on the ERS chILDEU CRC. RB reports consulting fees from Boehringer Ingelheim, Ferrer, and Sanofi and lecturer fees from Boehringer Ingelheim and Roche outside of the submitted work. RB also received support for attending meetings from Boehringer Ingelheim, Roche and Chiesi, and participation on advisory boards for Savara.
Figures
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
