

Accurate structure prediction of biomolecular interactions with AlphaFold 3
- PMID: 38718835
- PMCID: PMC11168924
- DOI: 10.1038/s41586-024-07487-w
Accurate structure prediction of biomolecular interactions with AlphaFold 3
Abstract
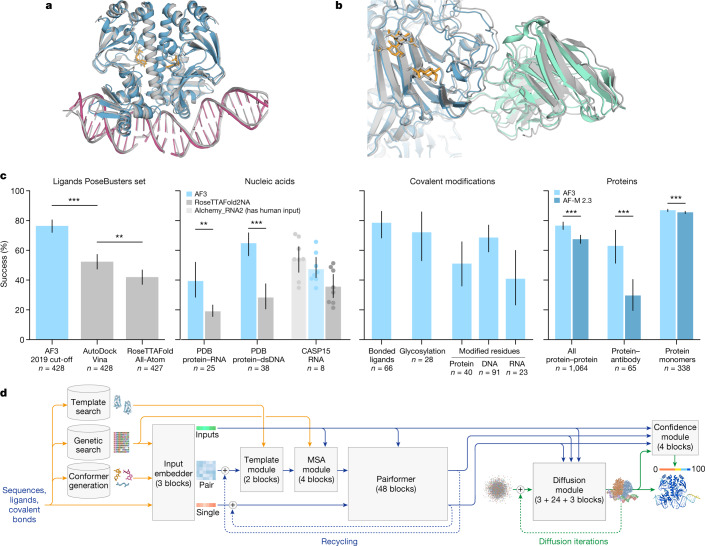
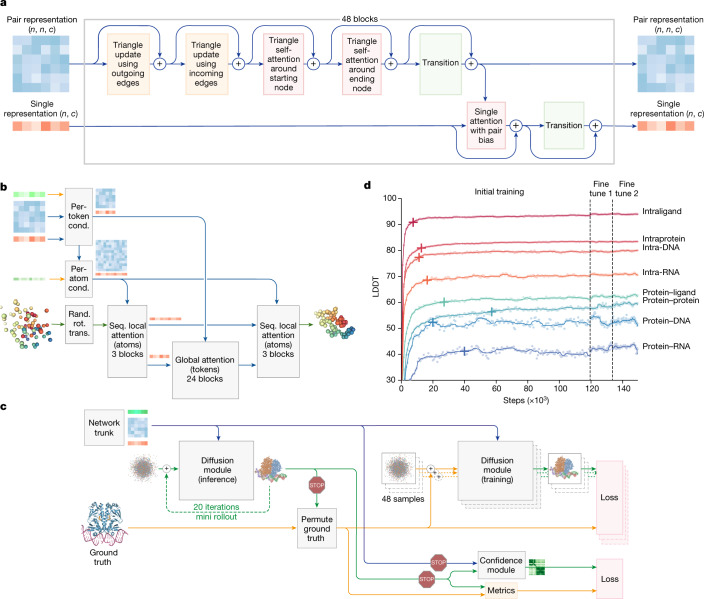
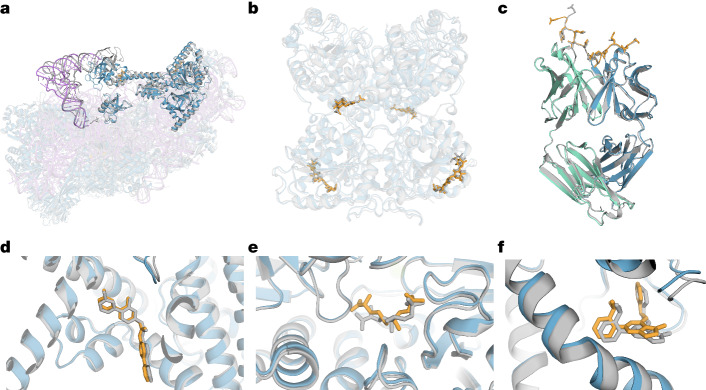
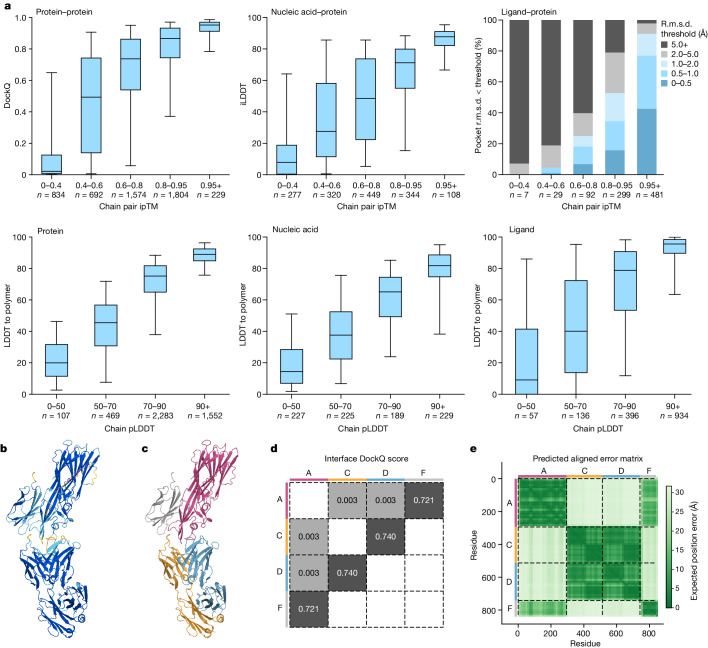
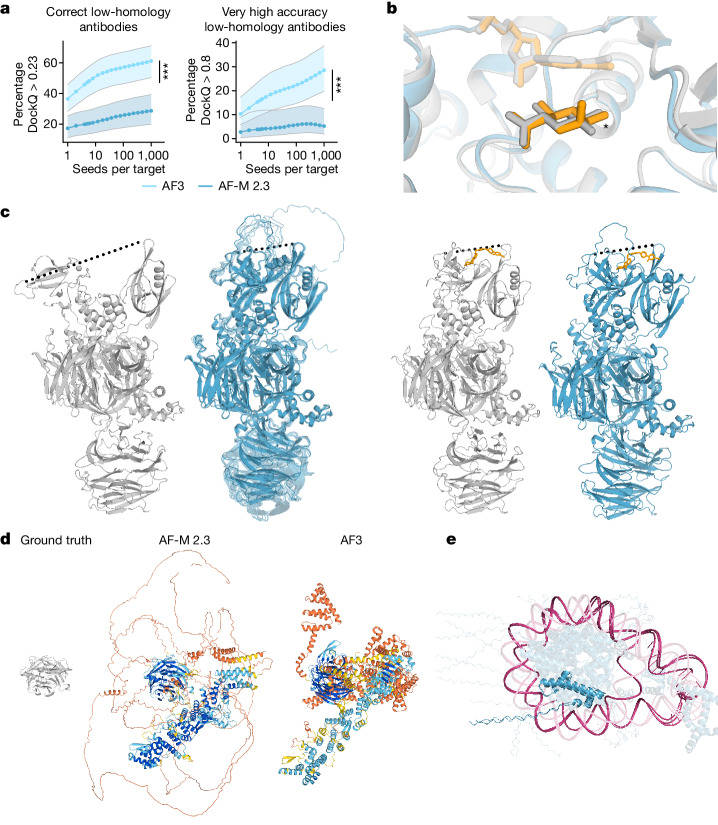
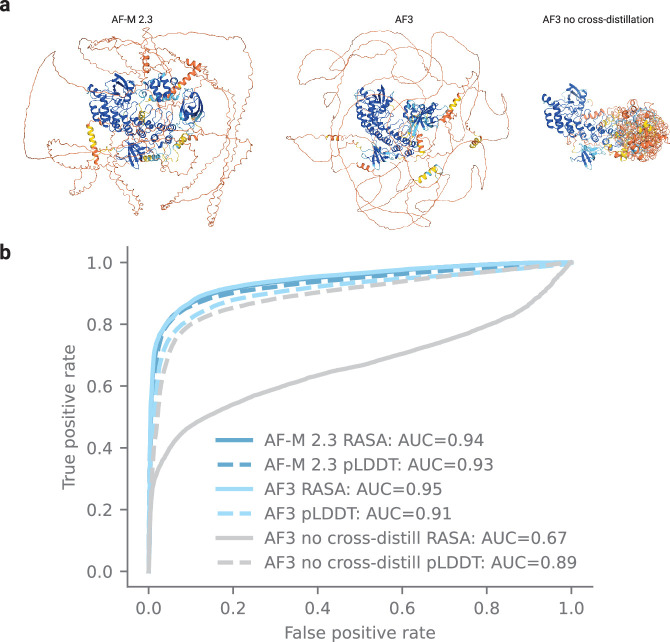
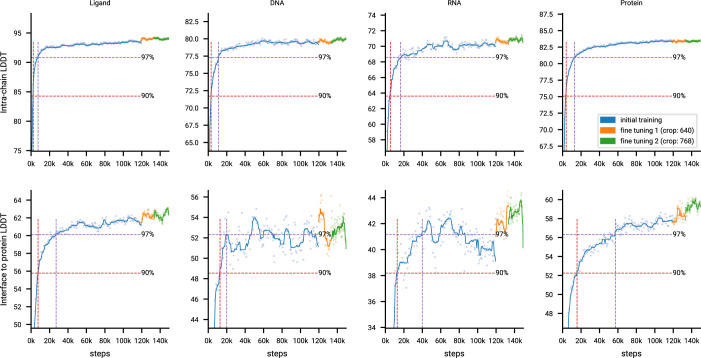
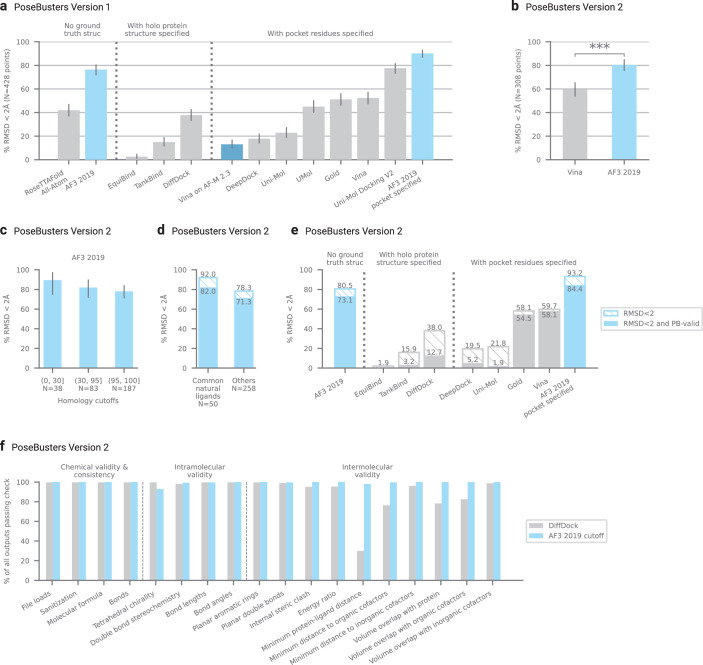
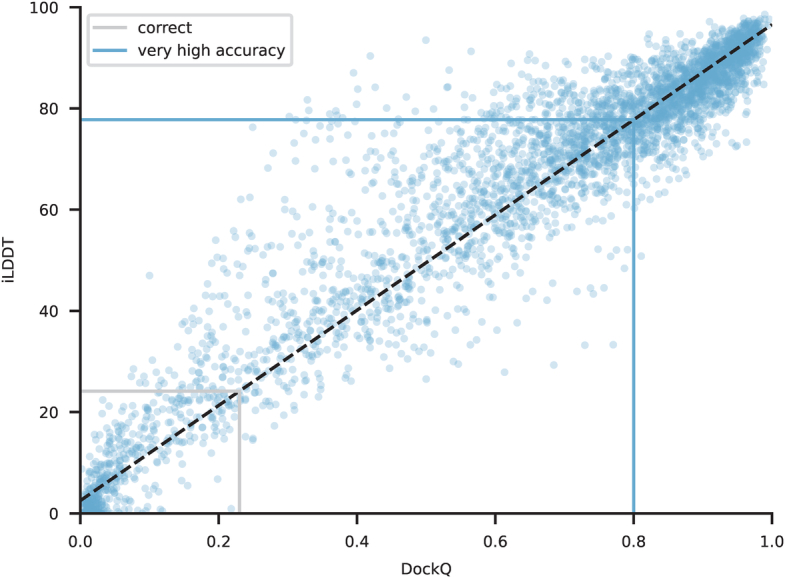
The introduction of AlphaFold 21 has spurred a revolution in modelling the structure of proteins and their interactions, enabling a huge range of applications in protein modelling and design2-6. Here we describe our AlphaFold 3 model with a substantially updated diffusion-based architecture that is capable of predicting the joint structure of complexes including proteins, nucleic acids, small molecules, ions and modified residues. The new AlphaFold model demonstrates substantially improved accuracy over many previous specialized tools: far greater accuracy for protein-ligand interactions compared with state-of-the-art docking tools, much higher accuracy for protein-nucleic acid interactions compared with nucleic-acid-specific predictors and substantially higher antibody-antigen prediction accuracy compared with AlphaFold-Multimer v.2.37,8. Together, these results show that high-accuracy modelling across biomolecular space is possible within a single unified deep-learning framework.
© 2024. The Author(s).
Conflict of interest statement
Author-affiliated entities have filed US provisional patent applications including 63/611,674, 63/611,638 and 63/546,444 relating to predicting 3D structures of molecule complexes using embedding neural networks and generative models. All of the authors other than A.B., Y.A.K. and E.D.Z. have commercial interests in the work described.
Figures

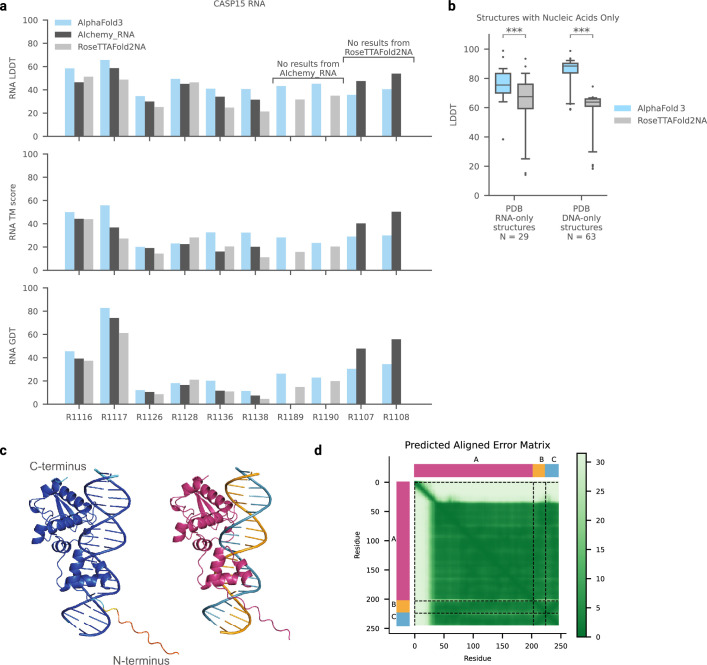
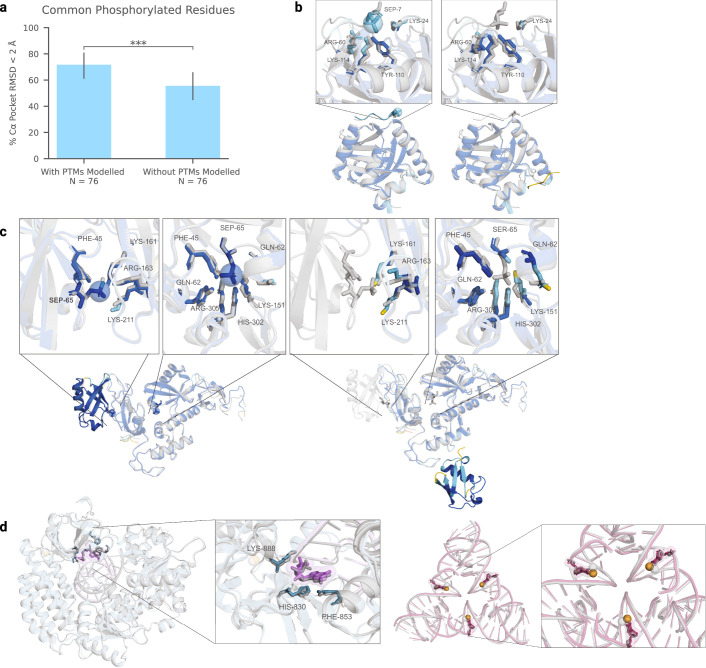
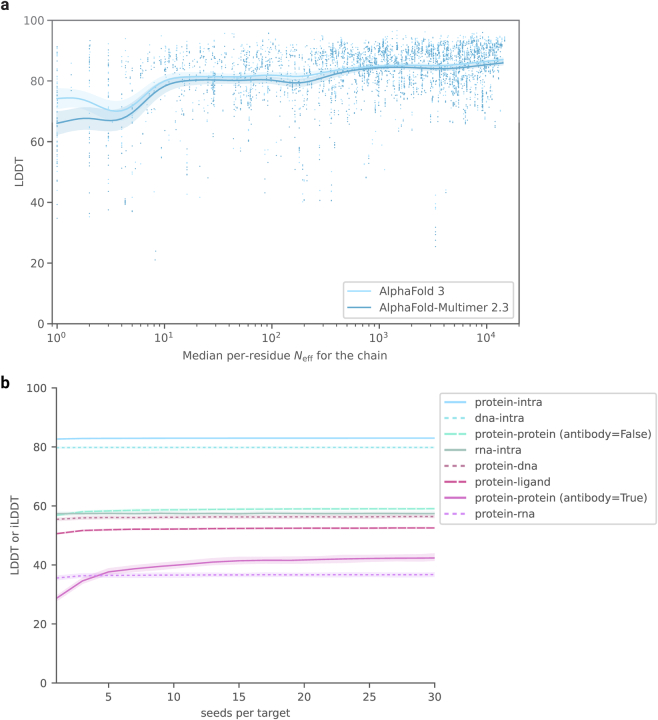
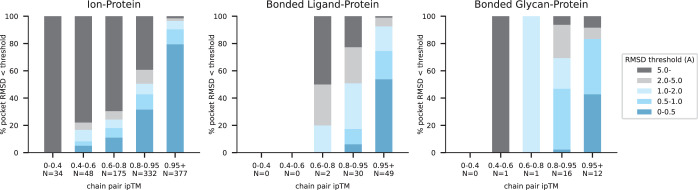
References
-
- Mosalaganti, S. et al. AI-based structure prediction empowers integrative structural analysis of human nuclear pores. Science376, eabm9506 (2022). - PubMed
-
- Anand, N. & Achim, T. Protein structure and sequence generation with equivariant denoising diffusion probabilistic models. Preprint at arXiv10.48550/arXiv.2205.15019 (2022).
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
