

Mapping genotypes to chromatin accessibility profiles in single cells
- PMID: 38720070
- PMCID: PMC11139586
- DOI: 10.1038/s41586-024-07388-y
Mapping genotypes to chromatin accessibility profiles in single cells
Abstract
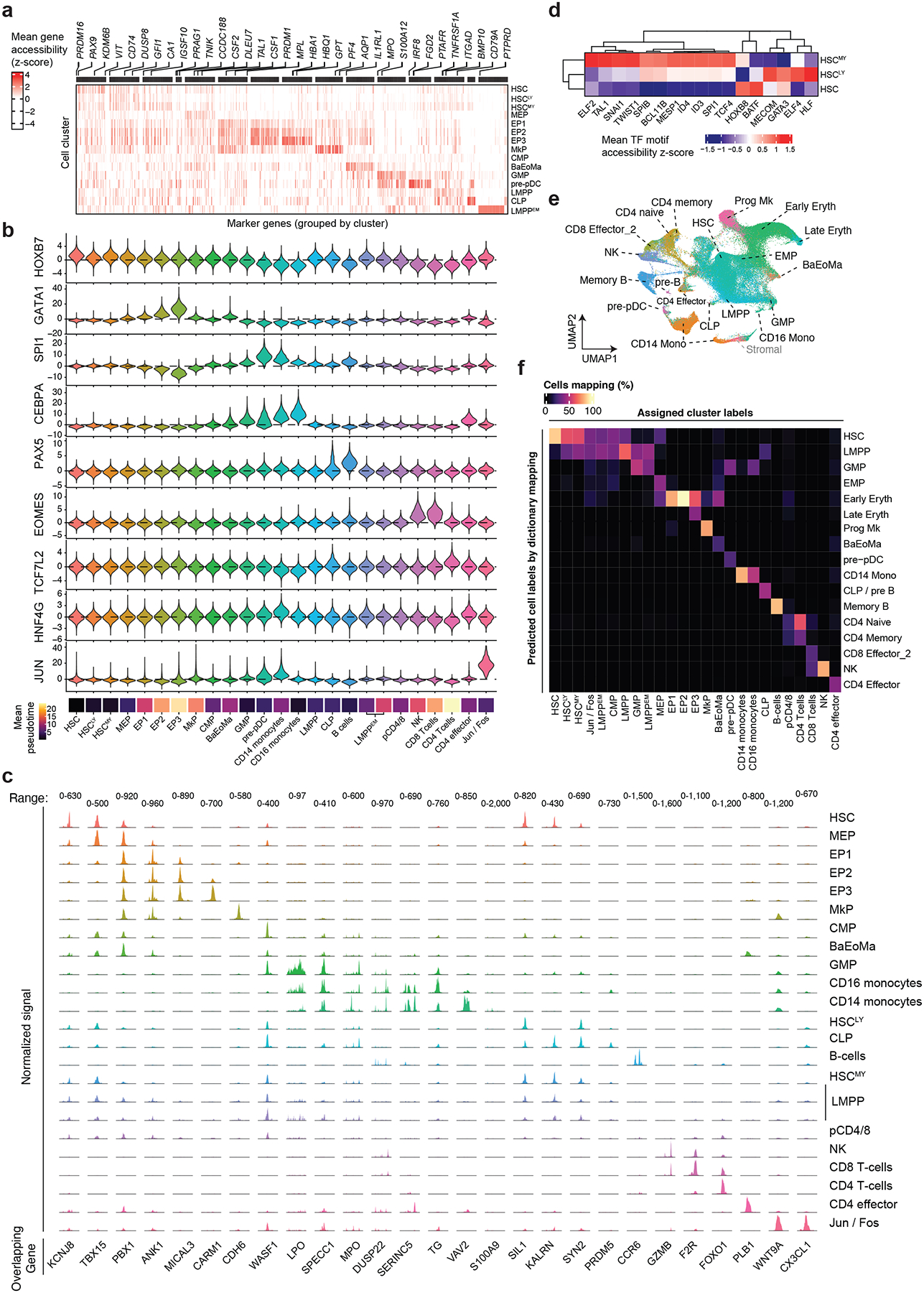
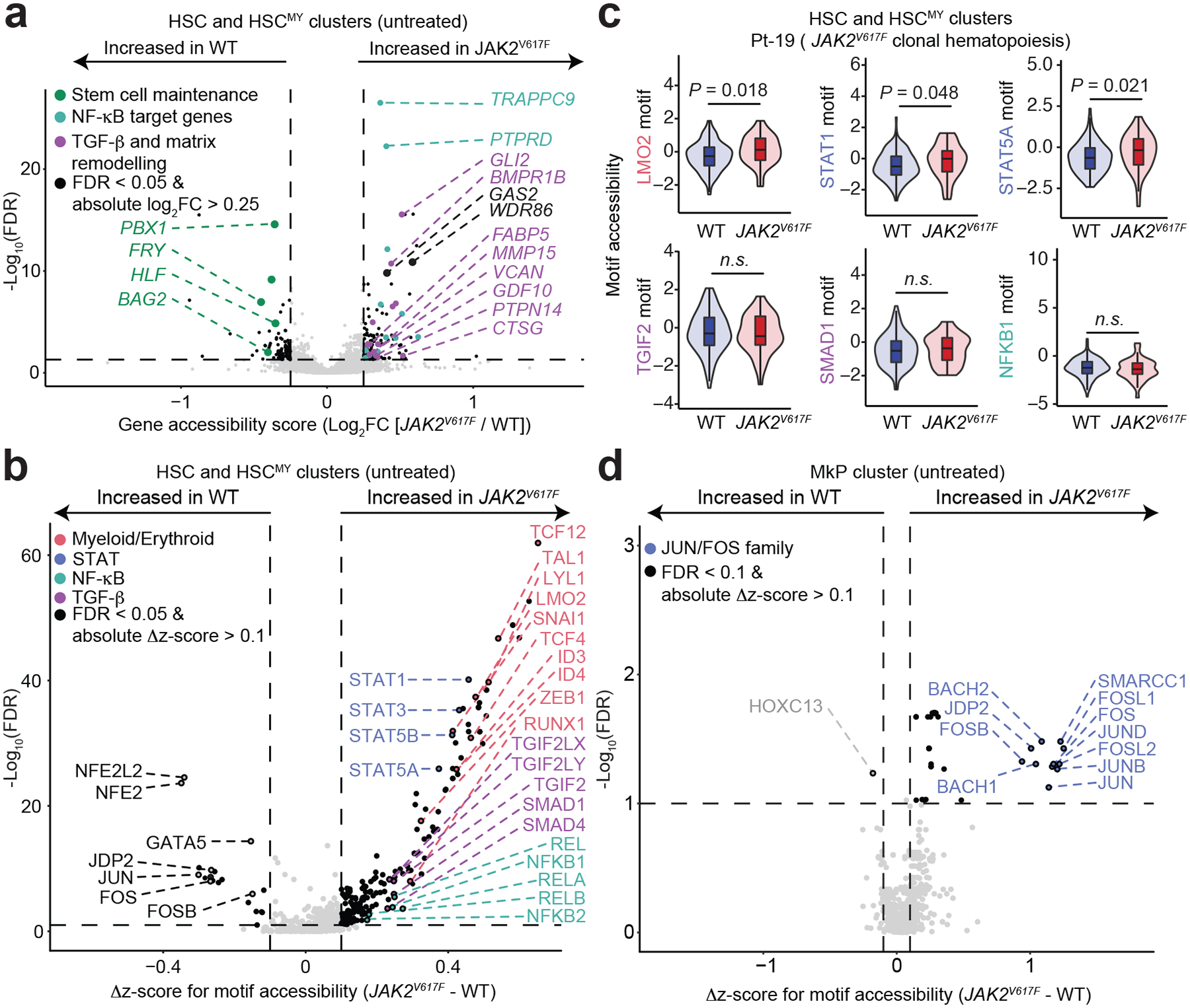
In somatic tissue differentiation, chromatin accessibility changes govern priming and precursor commitment towards cellular fates1-3. Therefore, somatic mutations are likely to alter chromatin accessibility patterns, as they disrupt differentiation topologies leading to abnormal clonal outgrowth. However, defining the impact of somatic mutations on the epigenome in human samples is challenging due to admixed mutated and wild-type cells. Here, to chart how somatic mutations disrupt epigenetic landscapes in human clonal outgrowths, we developed genotyping of targeted loci with single-cell chromatin accessibility (GoT-ChA). This high-throughput platform links genotypes to chromatin accessibility at single-cell resolution across thousands of cells within a single assay. We applied GoT-ChA to CD34+ cells from patients with myeloproliferative neoplasms with JAK2V617F-mutated haematopoiesis. Differential accessibility analysis between wild-type and JAK2V617F-mutant progenitors revealed both cell-intrinsic and cell-state-specific shifts within mutant haematopoietic precursors, including cell-intrinsic pro-inflammatory signatures in haematopoietic stem cells, and a distinct profibrotic inflammatory chromatin landscape in megakaryocytic progenitors. Integration of mitochondrial genome profiling and cell-surface protein expression measurement allowed expansion of genotyping onto DOGMA-seq through imputation, enabling single-cell capture of genotypes, chromatin accessibility, RNA expression and cell-surface protein expression. Collectively, we show that the JAK2V617F mutation leads to epigenetic rewiring in a cell-intrinsic and cell type-specific manner, influencing inflammation states and differentiation trajectories. We envision that GoT-ChA will empower broad future investigations of the critical link between somatic mutations and epigenetic alterations across clonal populations in malignant and non-malignant contexts.
© 2024. The Author(s), under exclusive licence to Springer Nature Limited.
Conflict of interest statement
M.S. served on the advisory board for Novartis, Kymera, Sierra Oncology, GSK, Rigel, BMS and Taiho; consulted for Boston Consulting and Dedham group and participated in GME activity for Novartis, Curis Oncology, Haymarket Media and Clinical care options. R.H. has served as a consultant for Protagonist Therapeutics, Inc., received research funding from Kartos Therapeutics, Inc., Novartis, and AbbVie Inc, and is on the Data Safety Monitoring Board of Novartis and AbbVie Inc. O.A.-W. has served as a consultant for H3B Biomedicine, Foundation Medicine Inc, Merck, Pfizer, Codify Therapeutics and Janssen, and is on the Scientific Advisory Board of Envisagenics Inc, AIChemy, and Codify Therapeutics. O.A.-W. has received prior research funding from H3B Biomedicine, LOXO Oncology, Nurix Therapeutics, Codify Therapeutics, and Minovia unrelated to the current manuscript. O.A.-W. is a scientific co-founder of Codify Therapeutics. P.S. and E.P.M. are current employees of 10x Genomics and Immunai, respectively. R.L.L. is on the supervisory board of Qiagen and is a scientific advisor to Imago, Mission Bio, Bakx, Zentalis, Ajax, Auron, Prelude, C4 Therapeutics and Isoplexis. R.L.L. has received research support from Abbvie, Constellation, Ajax, Zentalis and Prelude. R.L.L. has received research support from and consulted for Celgene and Roche and has consulted for Syndax, Incyte, Janssen, Astellas, Morphosys, and Novartis. R.L.L. has received honoraria from Astra Zeneca and Novartis for invited lectures and from Gilead and Novartis for grant reviews. D.A.L. has served as a consultant for Abbvie and Illumina and is on the Scientific Advisory Board of Mission Bio and C2i Genomics. D.A.L. has received prior research funding from BMS, 10x Genomics and Illumina unrelated to the current manuscript. R.M.M., F.I., E.P.M., R.C., P.S., and D.A.L. have filed a patent for GoT-ChA (#63/288,874). No other authors report competing interests.
Figures













References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
