

Genome organization around nuclear speckles drives mRNA splicing efficiency
- PMID: 38720076
- PMCID: PMC11164319
- DOI: 10.1038/s41586-024-07429-6
Genome organization around nuclear speckles drives mRNA splicing efficiency
Abstract
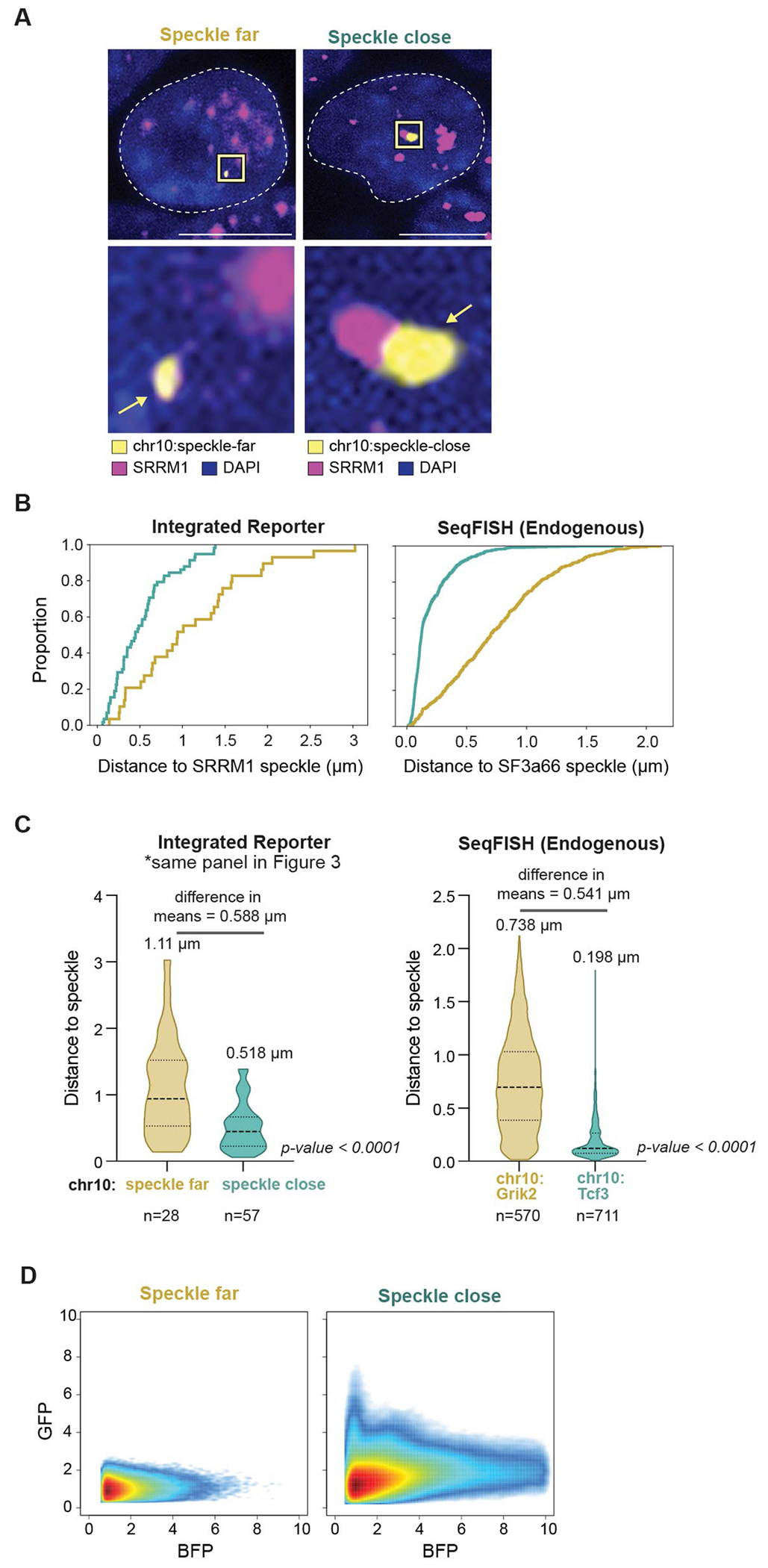
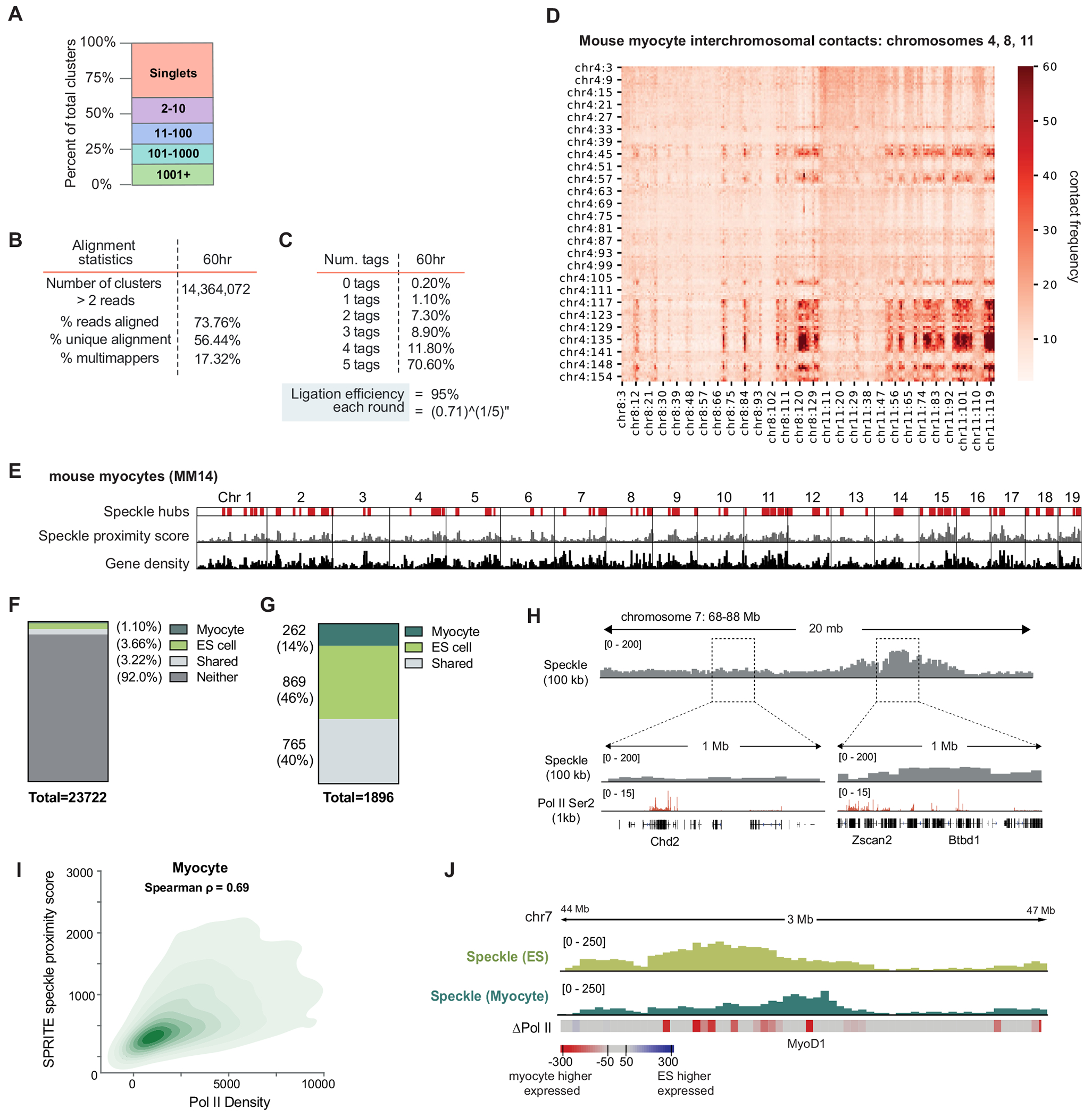
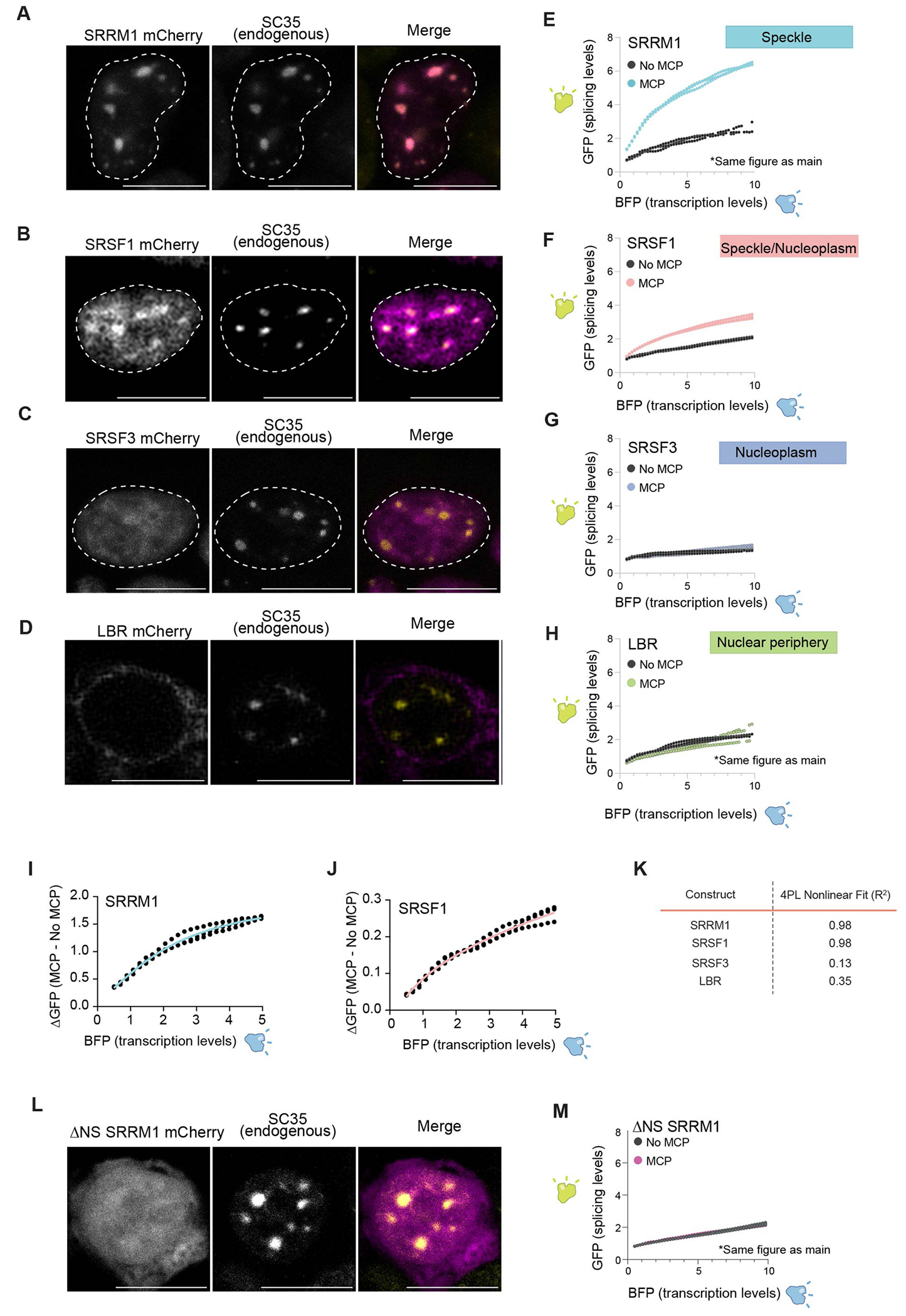
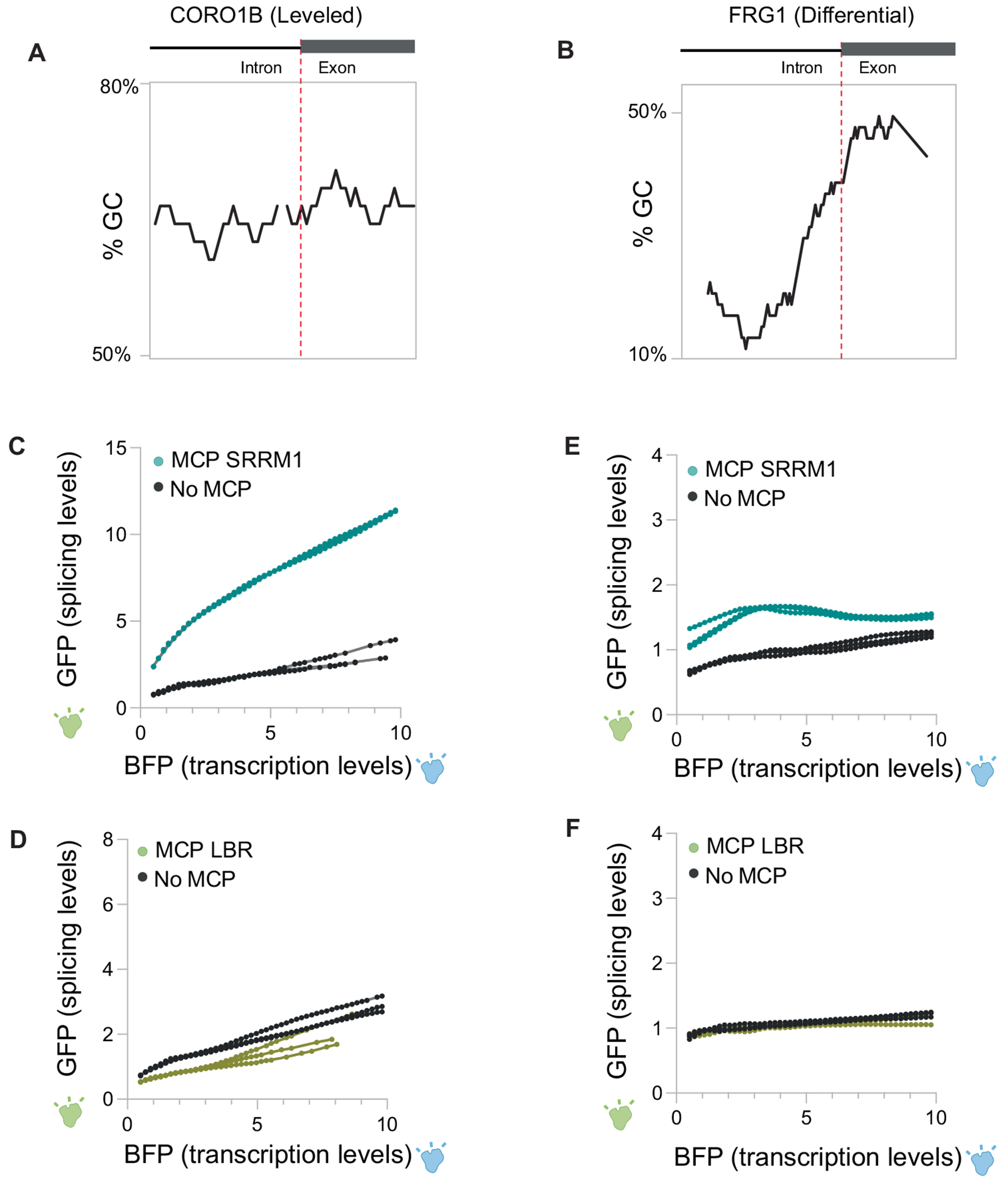
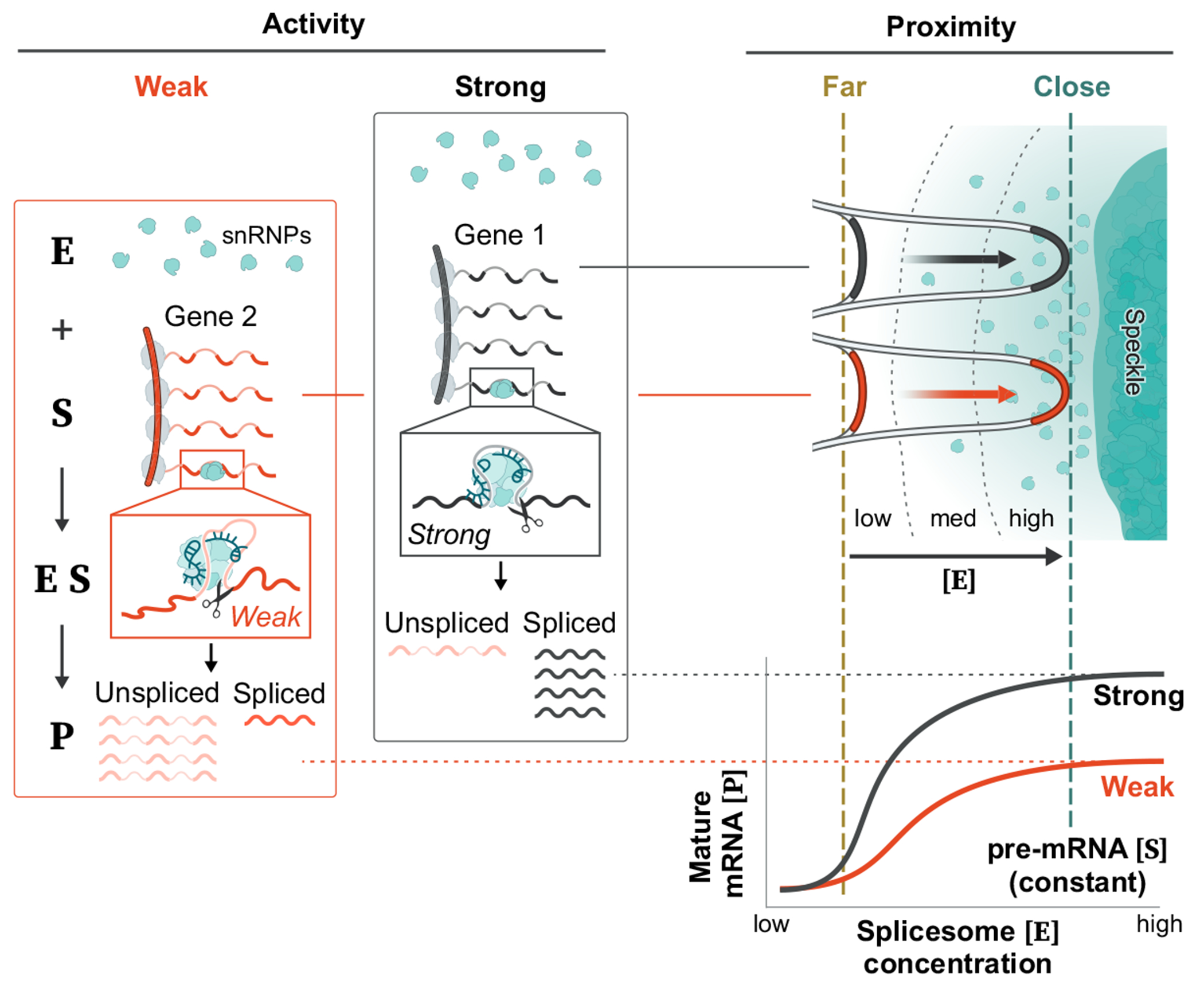
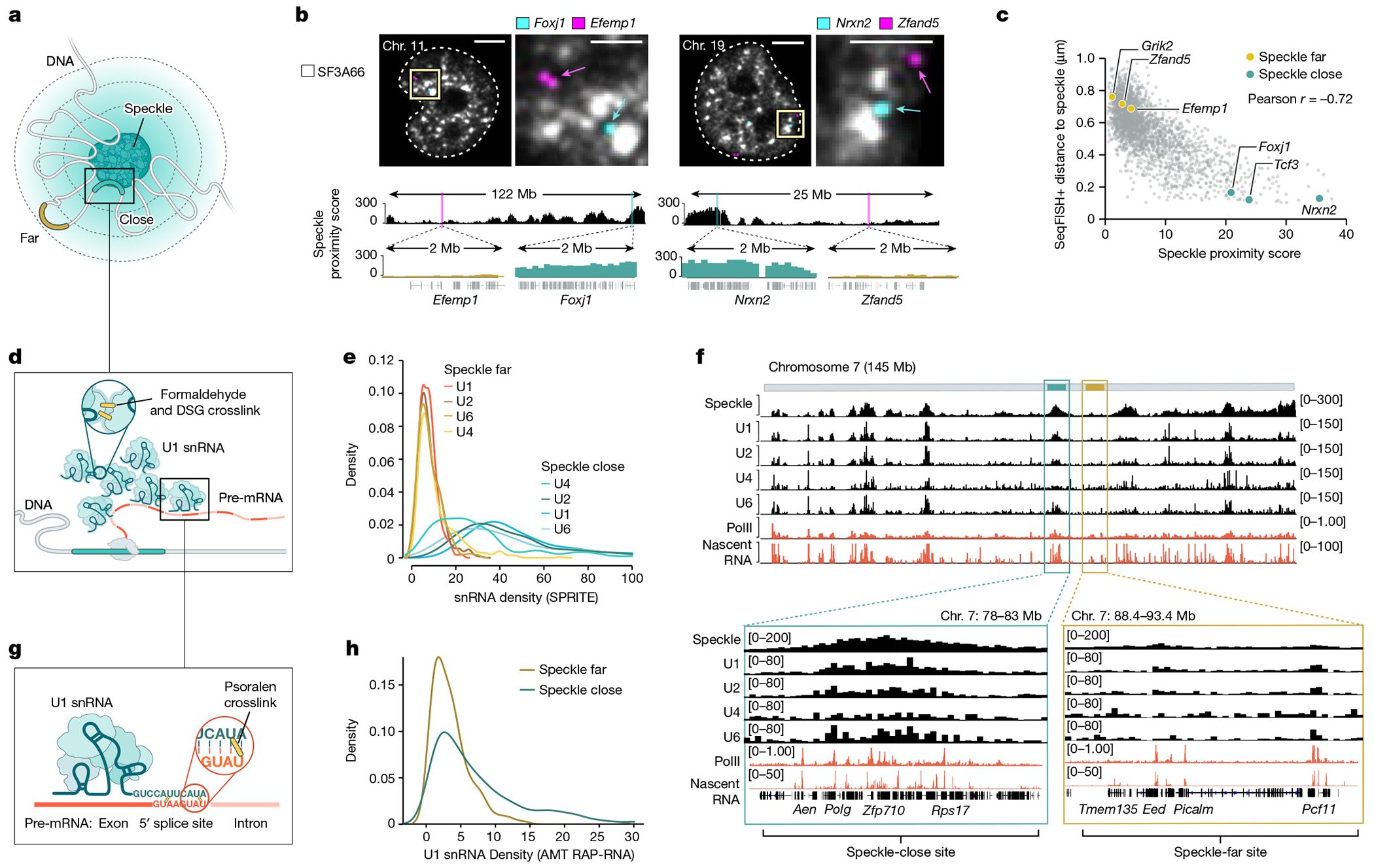
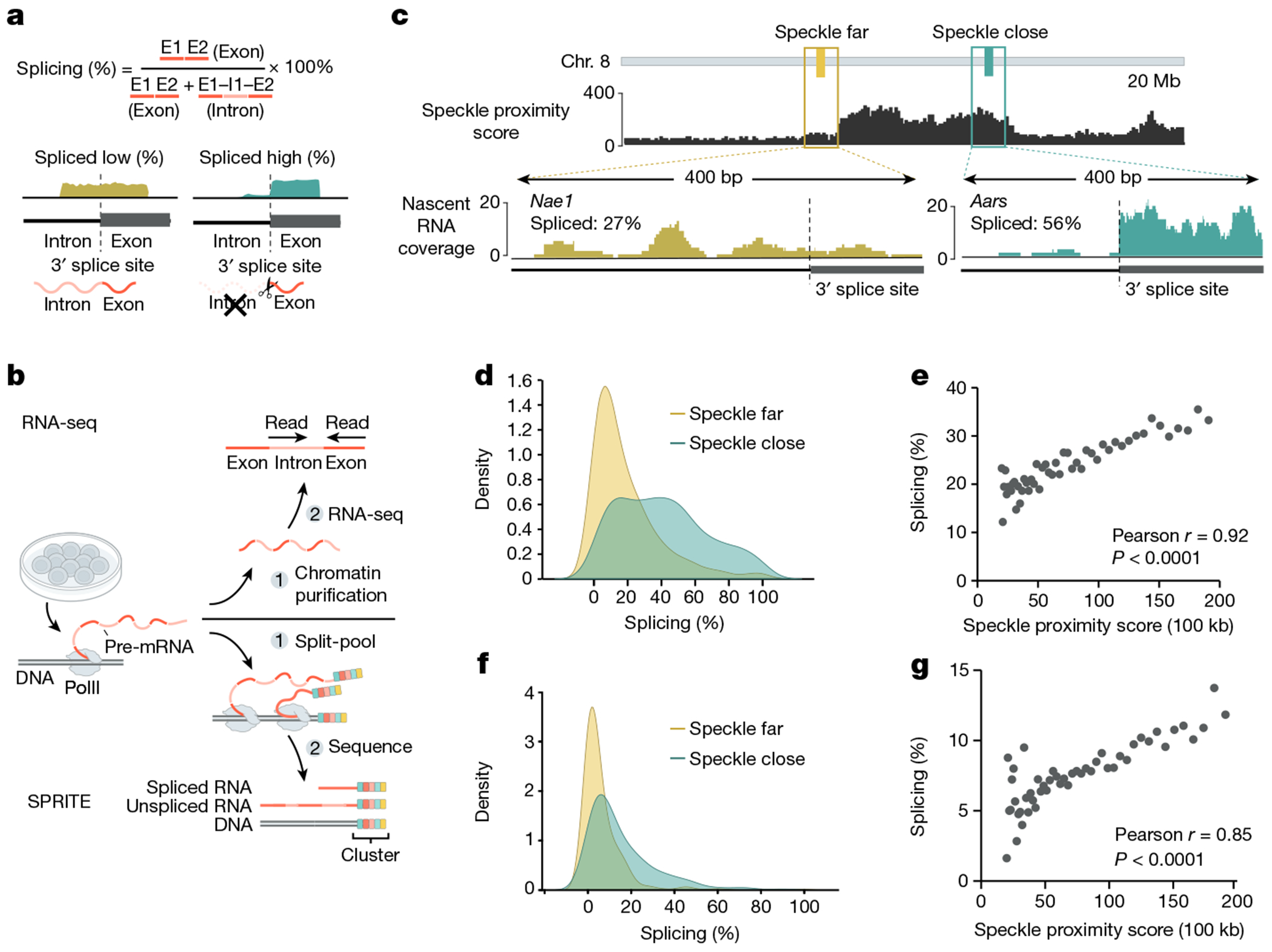
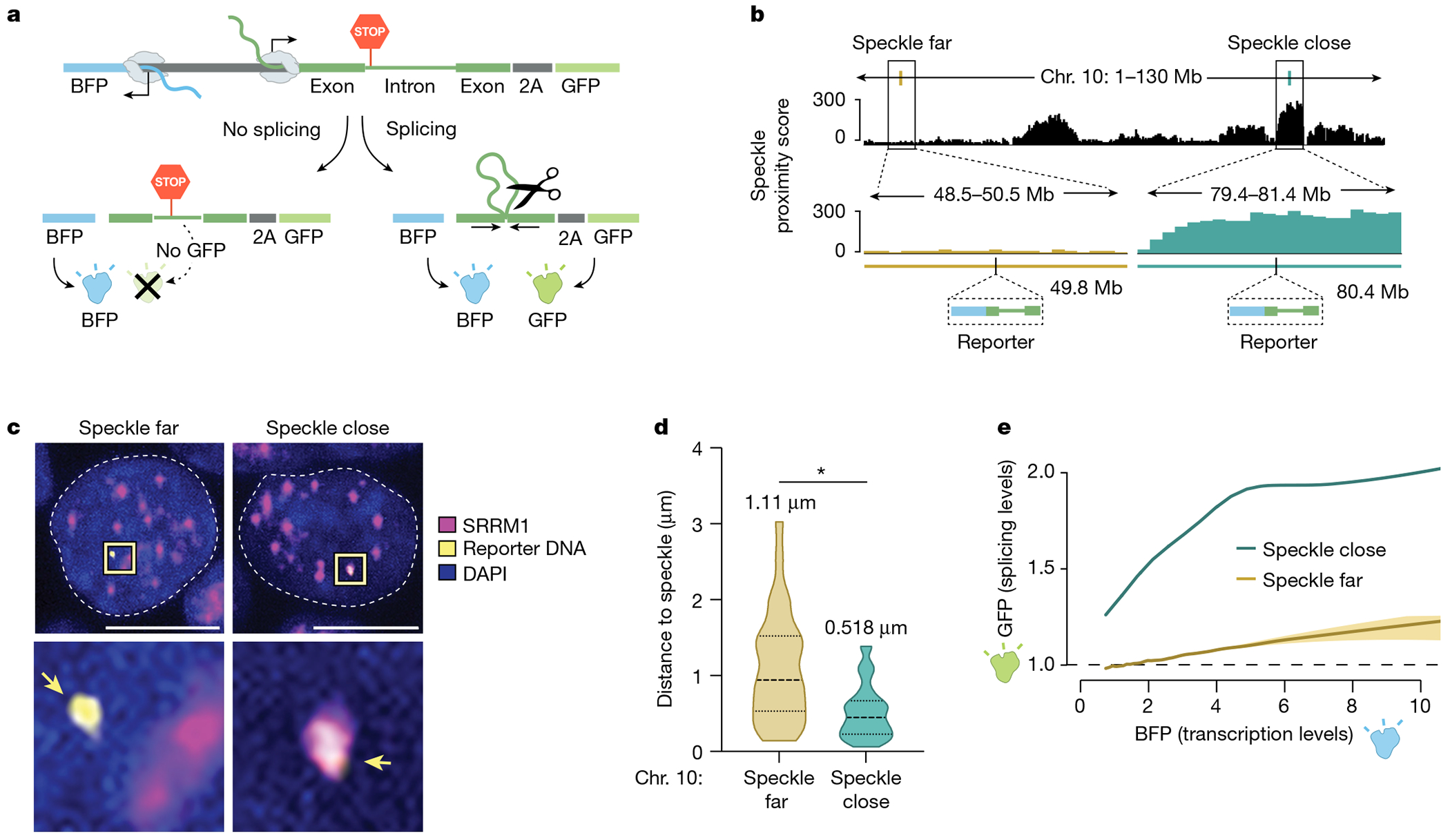
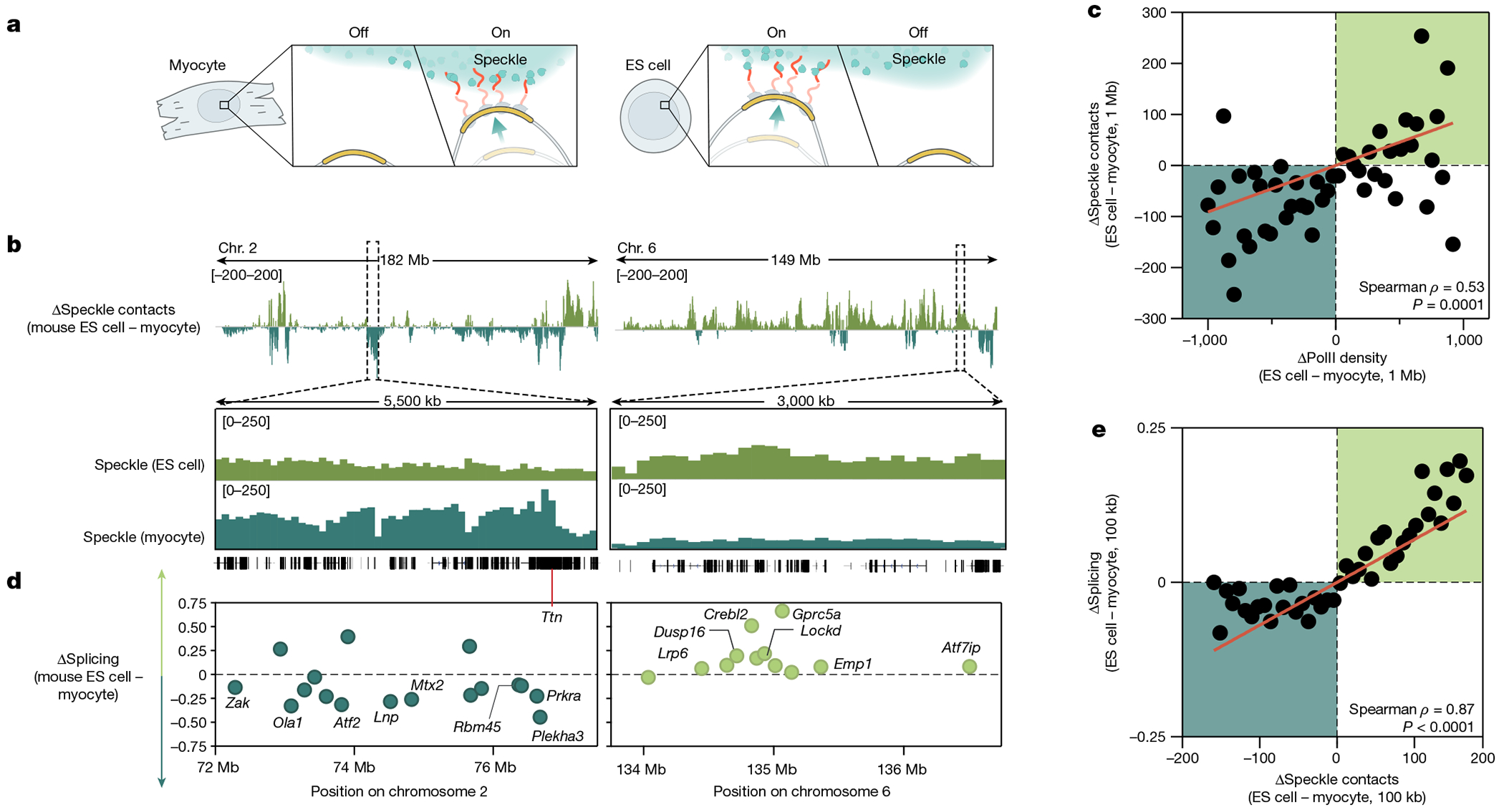
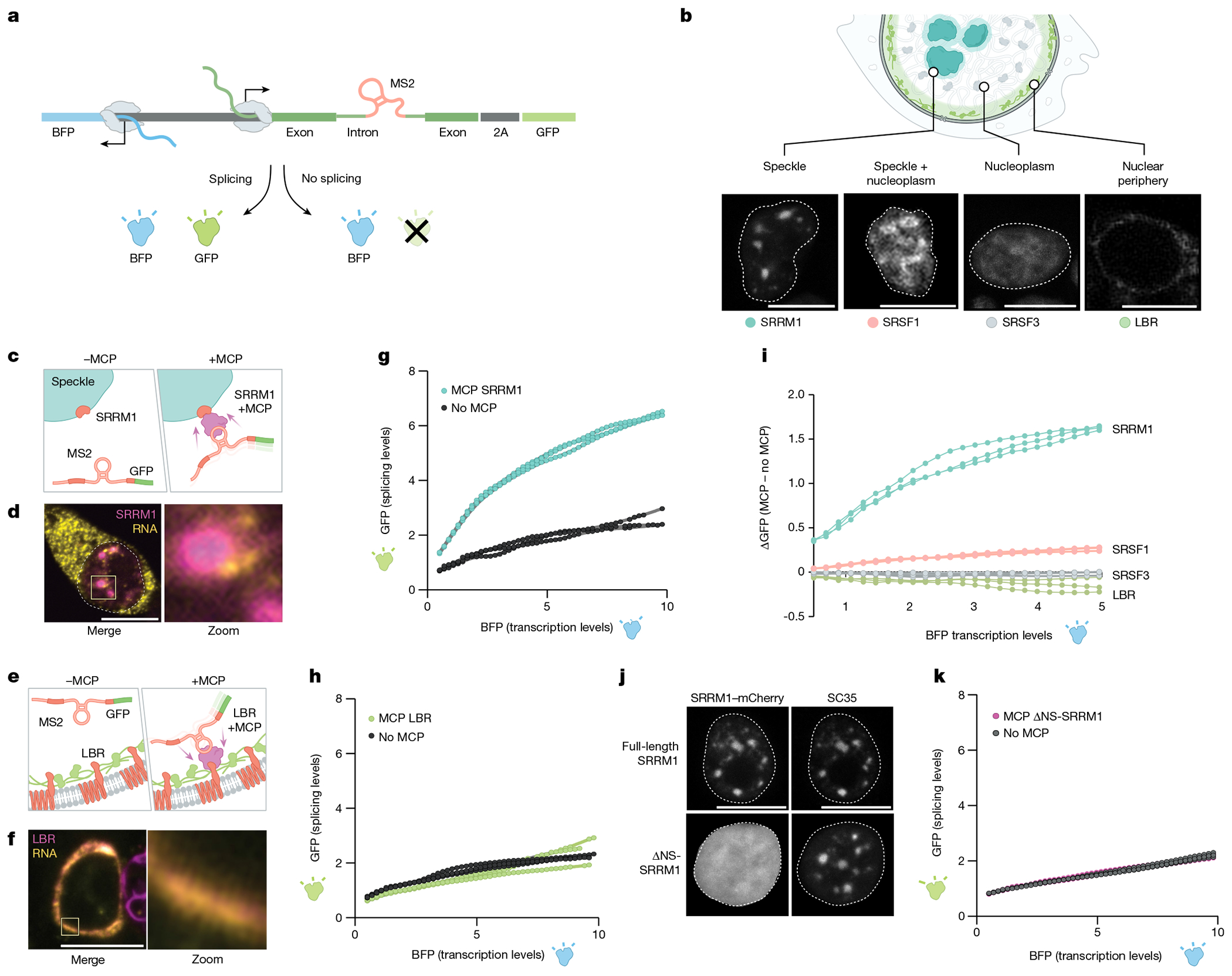
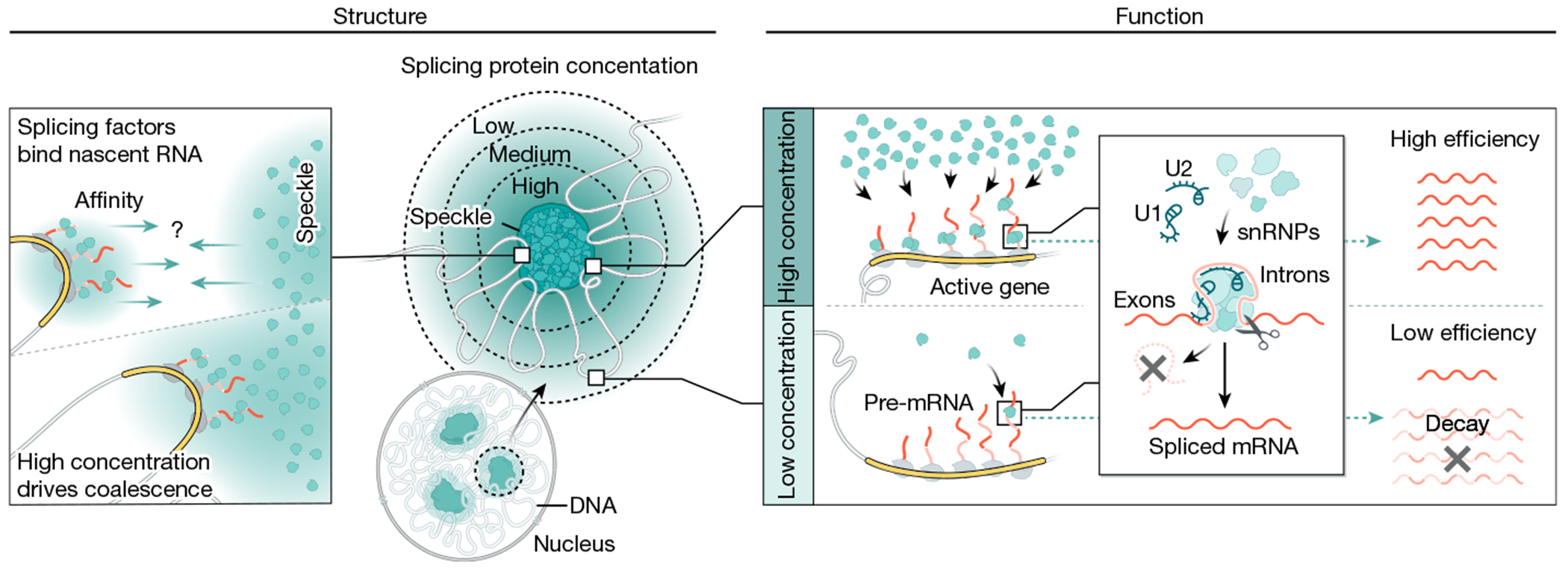
The nucleus is highly organized, such that factors involved in the transcription and processing of distinct classes of RNA are confined within specific nuclear bodies1,2. One example is the nuclear speckle, which is defined by high concentrations of protein and noncoding RNA regulators of pre-mRNA splicing3. What functional role, if any, speckles might play in the process of mRNA splicing is unclear4,5. Here we show that genes localized near nuclear speckles display higher spliceosome concentrations, increased spliceosome binding to their pre-mRNAs and higher co-transcriptional splicing levels than genes that are located farther from nuclear speckles. Gene organization around nuclear speckles is dynamic between cell types, and changes in speckle proximity lead to differences in splicing efficiency. Finally, directed recruitment of a pre-mRNA to nuclear speckles is sufficient to increase mRNA splicing levels. Together, our results integrate the long-standing observations of nuclear speckles with the biochemistry of mRNA splicing and demonstrate a crucial role for dynamic three-dimensional spatial organization of genomic DNA in driving spliceosome concentrations and controlling the efficiency of mRNA splicing.
© 2024. The Author(s), under exclusive licence to Springer Nature Limited.
Conflict of interest statement
Figures
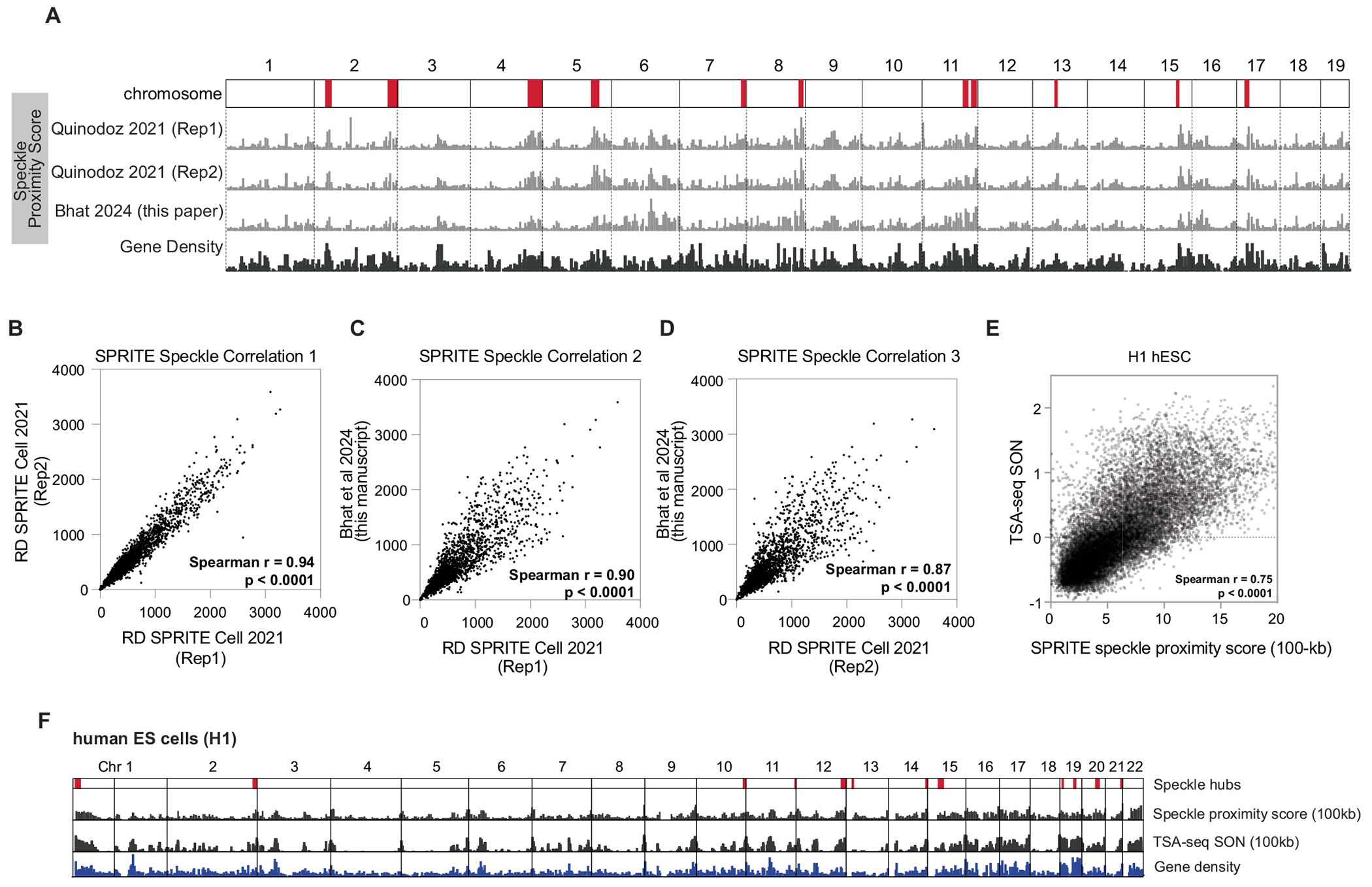
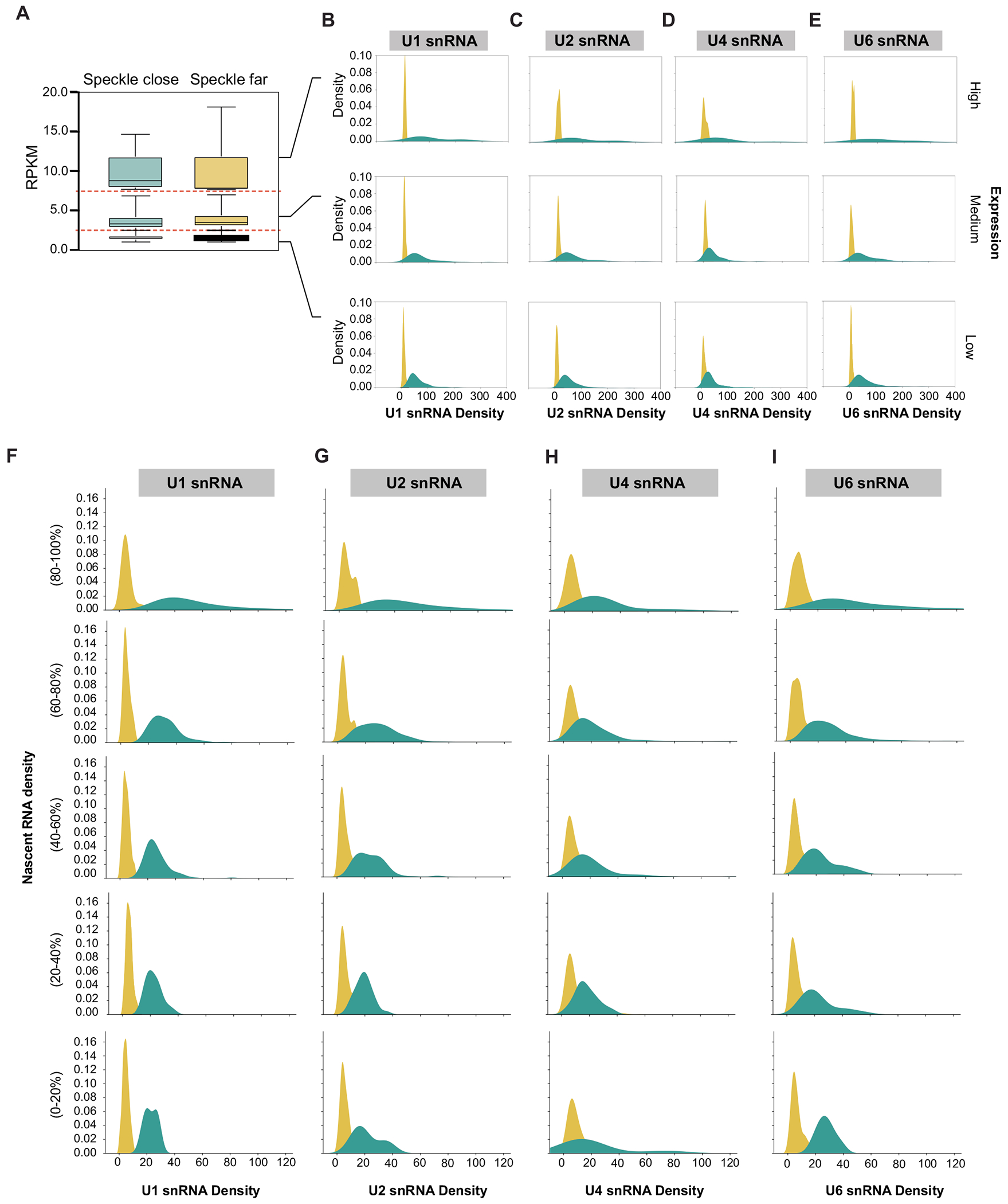
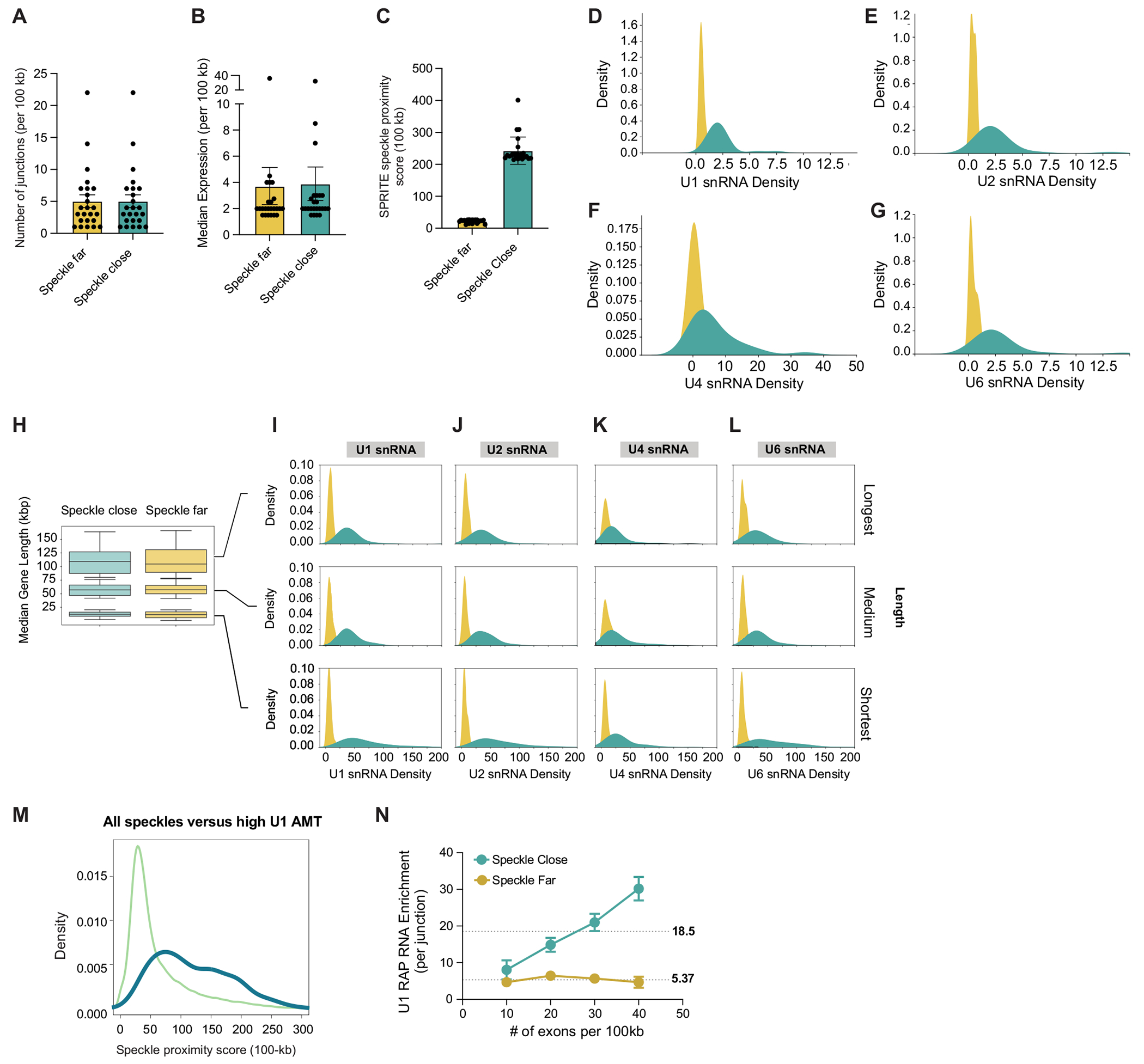
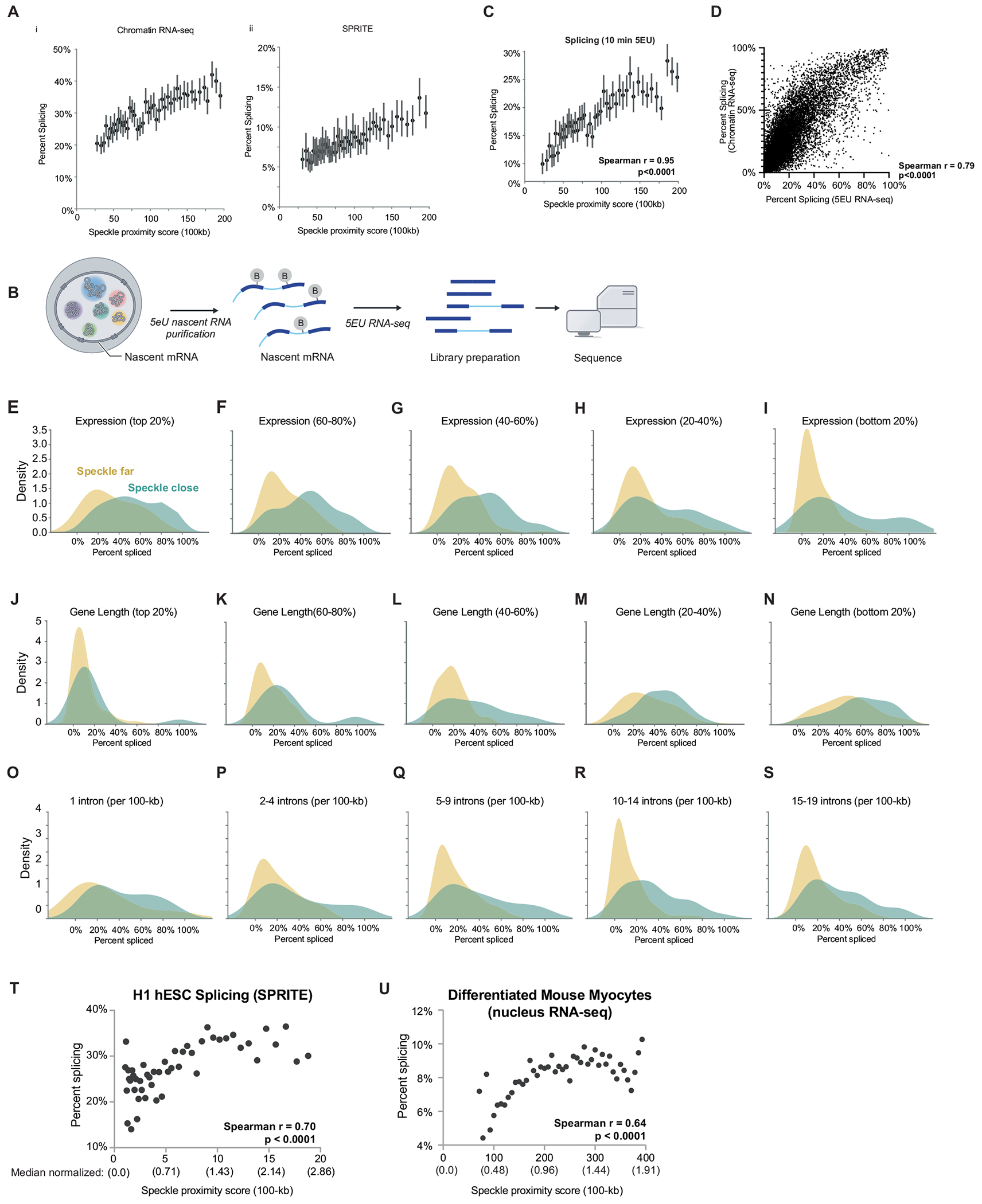
Update of
-
3D genome organization around nuclear speckles drives mRNA splicing efficiency.bioRxiv [Preprint]. 2023 Jan 4:2023.01.04.522632. doi: 10.1101/2023.01.04.522632. bioRxiv. 2023. Update in: Nature. 2024 May;629(8014):1165-1173. doi: 10.1038/s41586-024-07429-6. PMID: 36711853 Free PMC article. Updated. Preprint.
References
-
- Bhat P, Honson D & Guttman M Nuclear compartmentalization as a mechanism of quantitative control of gene expression. Nat. Rev. Mol. Cell Biol 22, 653–670 (2021). - PubMed
-
- Lewis JD & Tollervey D Like attracts like: getting RNA processing together in the nucleus. Science 288, 1385–1389 (2000). - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
