

Mitochondria regulate proliferation in adult cardiac myocytes
- PMID: 38722697
- PMCID: PMC11213516
- DOI: 10.1172/JCI165482
Mitochondria regulate proliferation in adult cardiac myocytes
Abstract
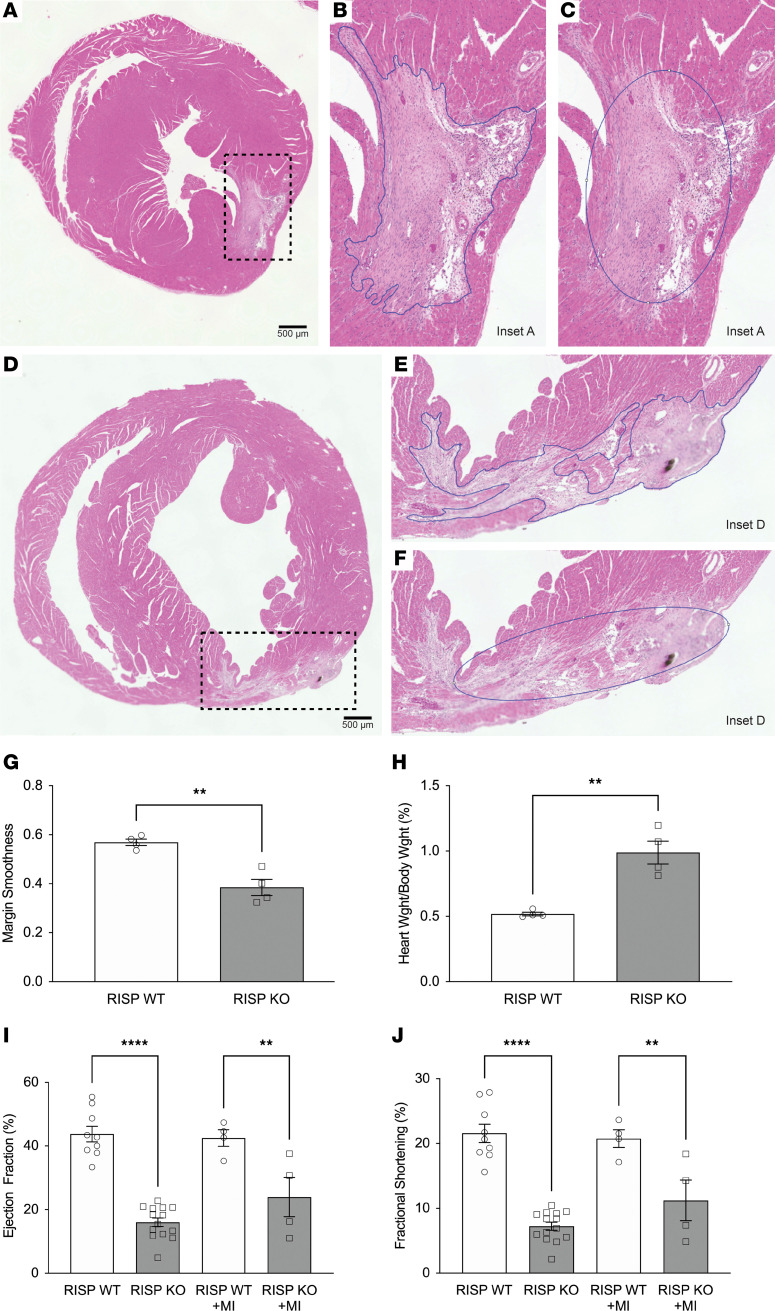
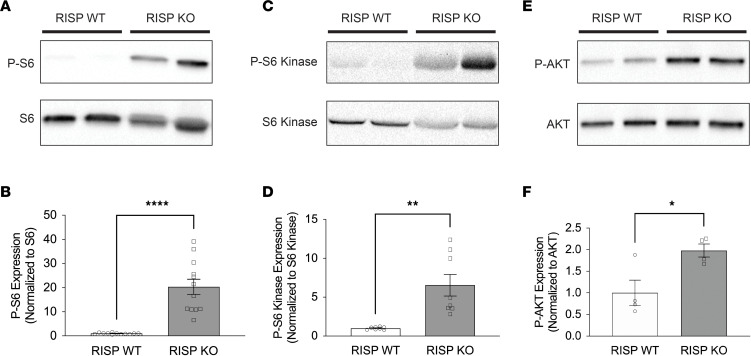
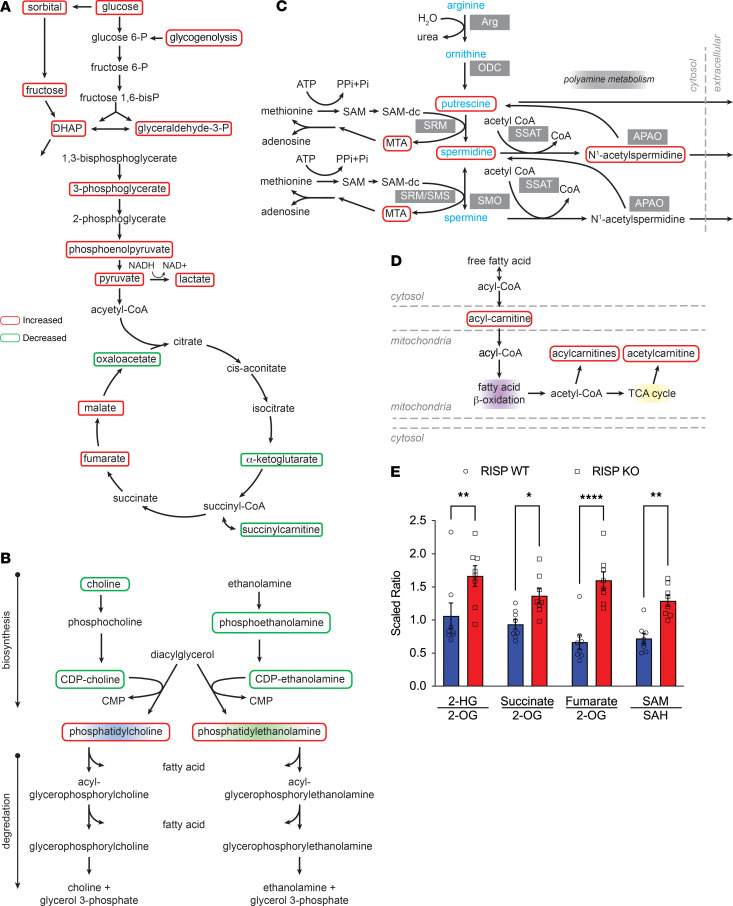
Newborn mammalian cardiomyocytes quickly transition from a fetal to an adult phenotype that utilizes mitochondrial oxidative phosphorylation but loses mitotic capacity. We tested whether forced reversal of adult cardiomyocytes back to a fetal glycolytic phenotype would restore proliferative capacity. We deleted Uqcrfs1 (mitochondrial Rieske iron-sulfur protein, RISP) in hearts of adult mice. As RISP protein decreased, heart mitochondrial function declined, and glucose utilization increased. Simultaneously, the hearts underwent hyperplastic remodeling during which cardiomyocyte number doubled without cellular hypertrophy. Cellular energy supply was preserved, AMPK activation was absent, and mTOR activation was evident. In ischemic hearts with RISP deletion, new cardiomyocytes migrated into the infarcted region, suggesting the potential for therapeutic cardiac regeneration. RNA sequencing revealed upregulation of genes associated with cardiac development and proliferation. Metabolomic analysis revealed a decrease in α-ketoglutarate (required for TET-mediated demethylation) and an increase in S-adenosylmethionine (required for methyltransferase activity). Analysis revealed an increase in methylated CpGs near gene transcriptional start sites. Genes that were both differentially expressed and differentially methylated were linked to upregulated cardiac developmental pathways. We conclude that decreased mitochondrial function and increased glucose utilization can restore mitotic capacity in adult cardiomyocytes, resulting in the generation of new heart cells, potentially through the modification of substrates that regulate epigenetic modification of genes required for proliferation.
Keywords: Bioenergetics; Cardiology; Cardiovascular disease; Metabolism; Mitochondria.
Conflict of interest statement
Figures







References
MeSH terms
Substances
Grants and funding
- P01 AG049665/AG/NIA NIH HHS/United States
- R01 HL122309/HL/NHLBI NIH HHS/United States
- P30 CA060553/CA/NCI NIH HHS/United States
- R35 HL155652/HL/NHLBI NIH HHS/United States
- R01 HL147575/HL/NHLBI NIH HHS/United States
- R01 HL149883/HL/NHLBI NIH HHS/United States
- R01 HL122062/HL/NHLBI NIH HHS/United States
- R01 HL153122/HL/NHLBI NIH HHS/United States
- R01 HL109478/HL/NHLBI NIH HHS/United States
- T32 DK007169/DK/NIDDK NIH HHS/United States
- R01 HL118491/HL/NHLBI NIH HHS/United States
- P01 HL154998/HL/NHLBI NIH HHS/United States
- R01 HL035440/HL/NHLBI NIH HHS/United States
- R01 HL159964/HL/NHLBI NIH HHS/United States
- R01 HL147290/HL/NHLBI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
