

Systematic epigenome editing captures the context-dependent instructive function of chromatin modifications
- PMID: 38724747
- PMCID: PMC11176084
- DOI: 10.1038/s41588-024-01706-w
Systematic epigenome editing captures the context-dependent instructive function of chromatin modifications
Abstract
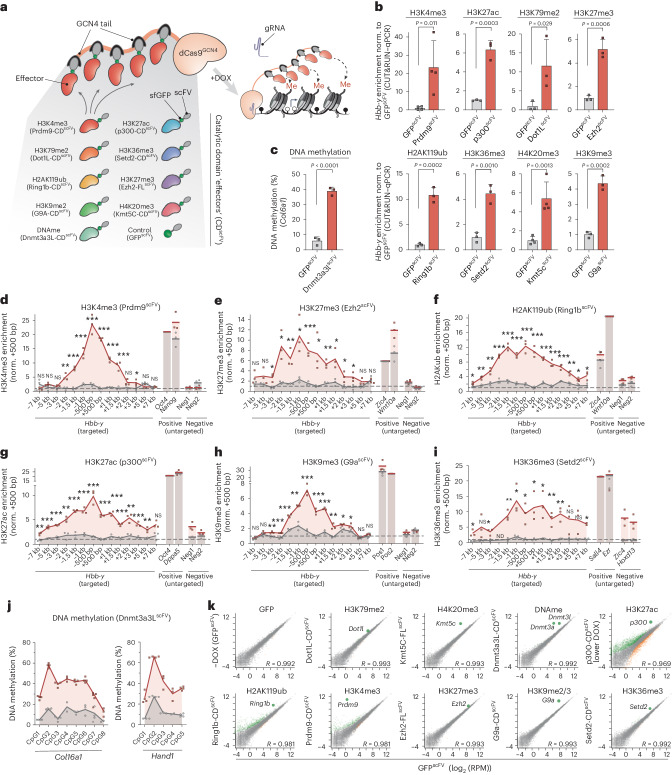
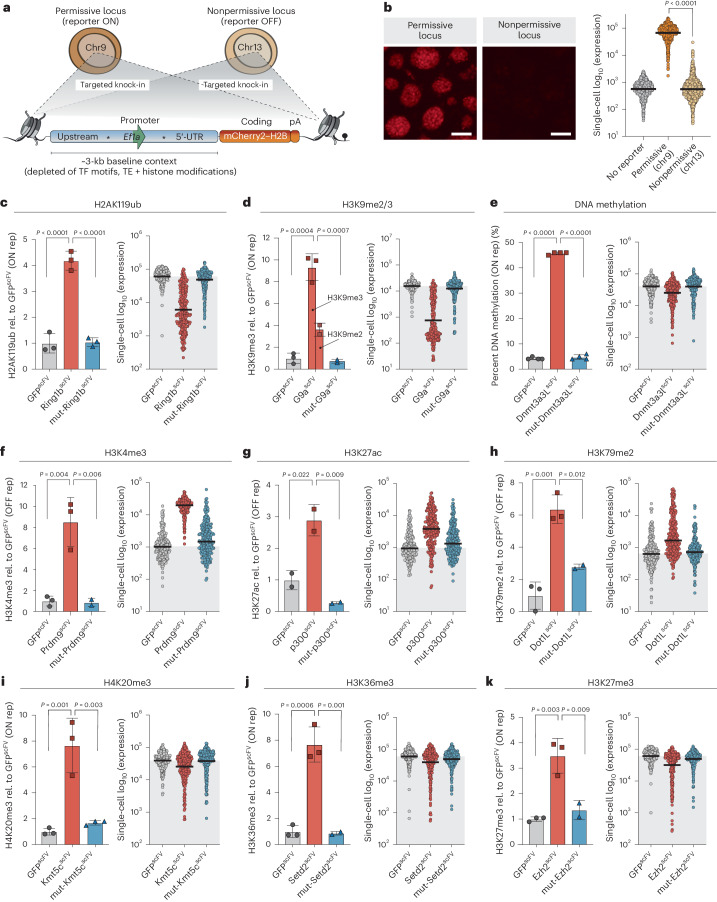
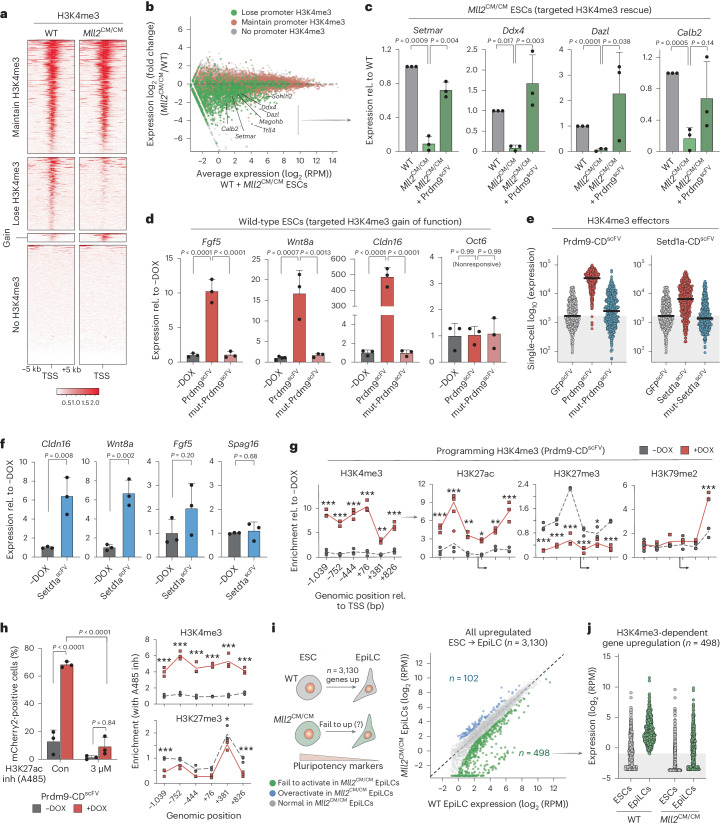
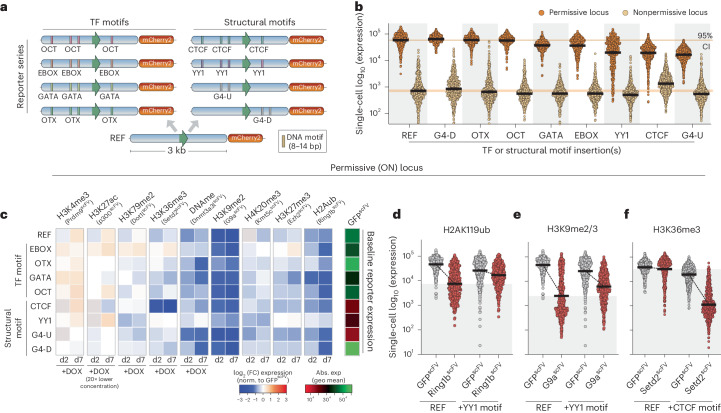
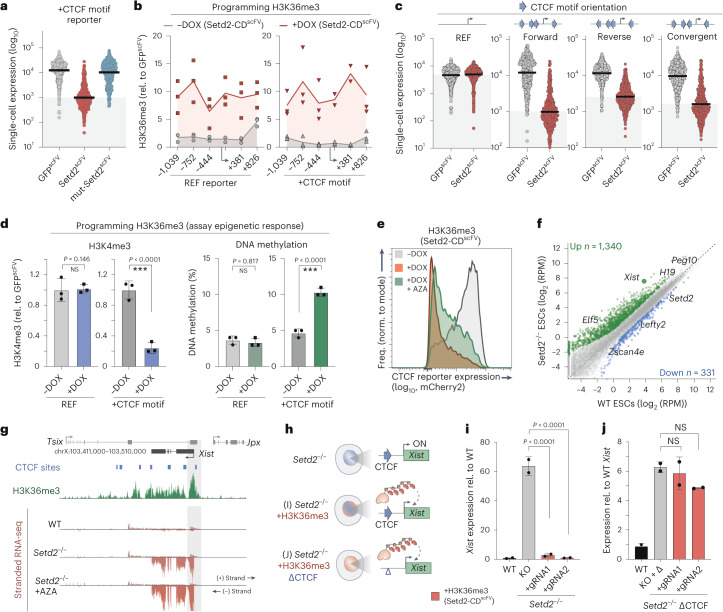
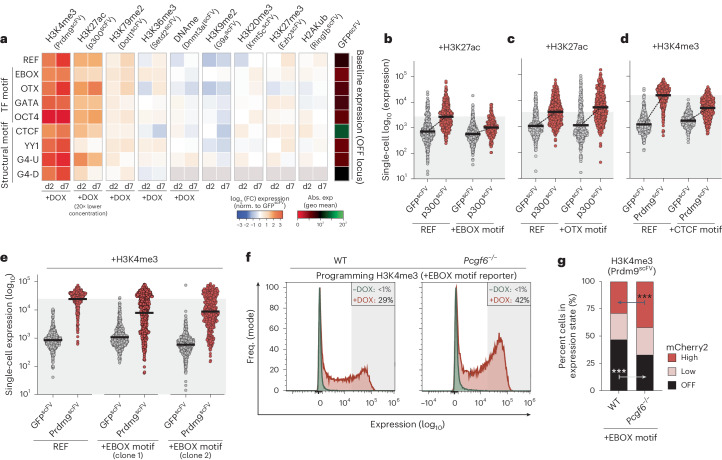
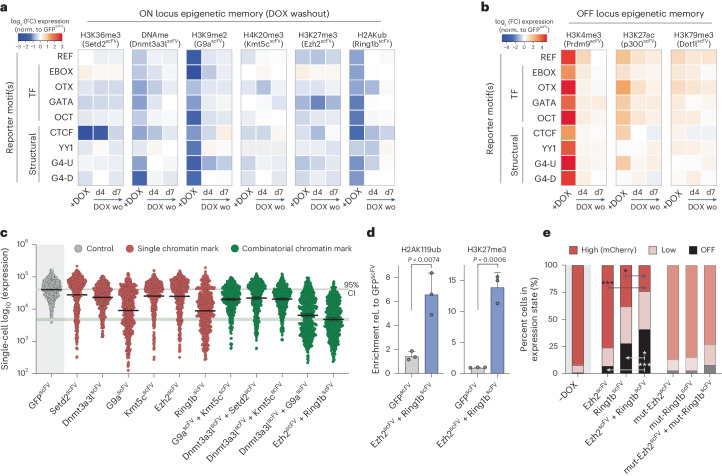
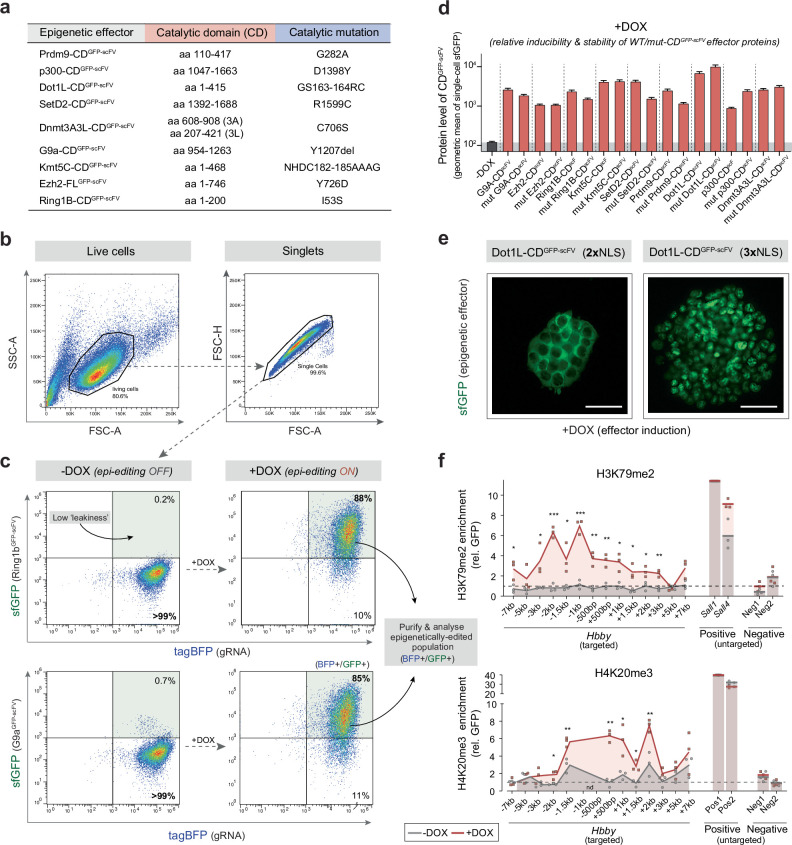
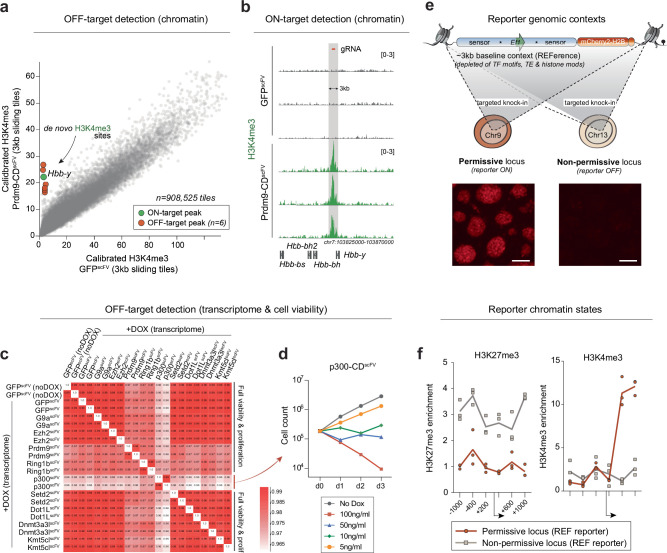
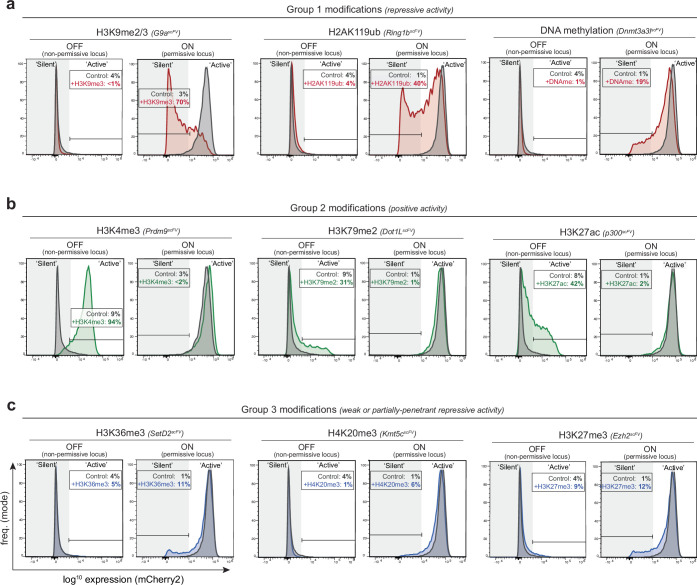
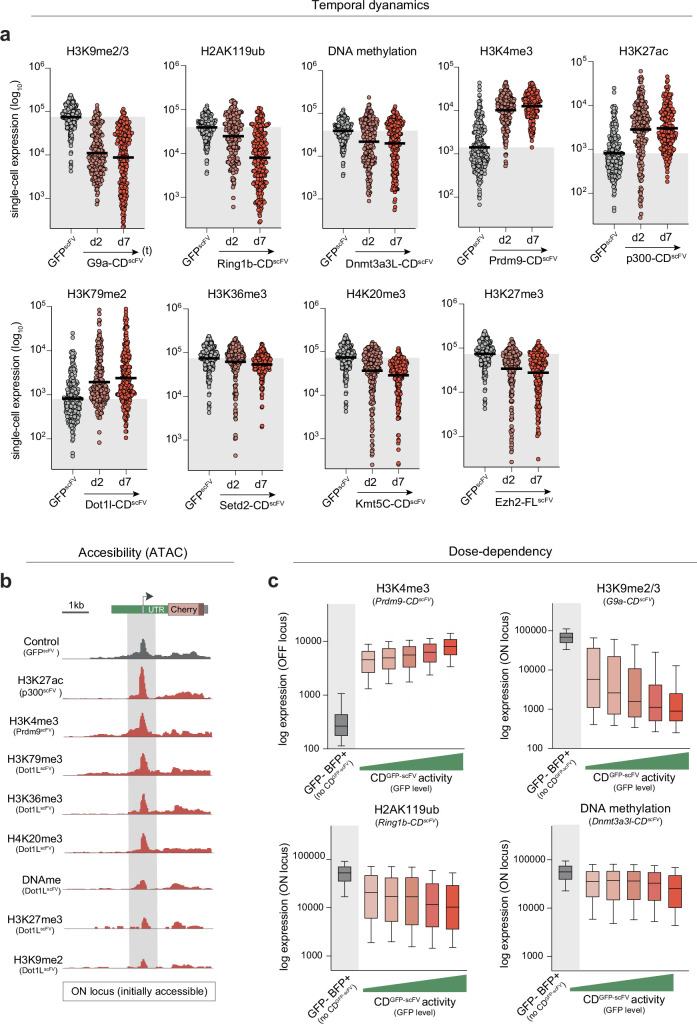
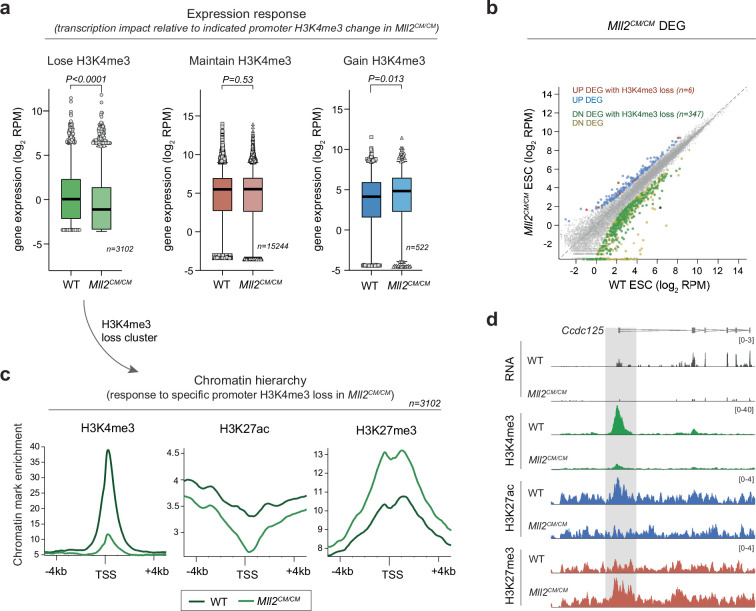
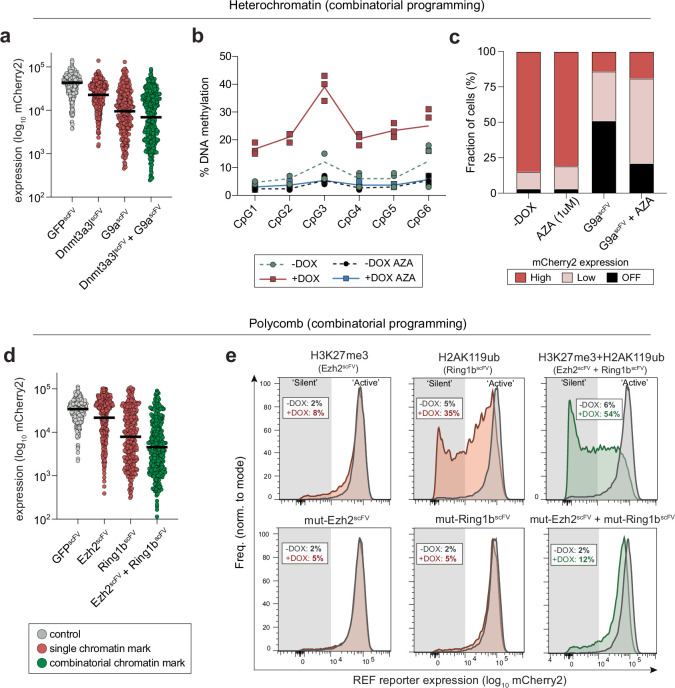
Chromatin modifications are linked with regulating patterns of gene expression, but their causal role and context-dependent impact on transcription remains unresolved. Here we develop a modular epigenome editing platform that programs nine key chromatin modifications, or combinations thereof, to precise loci in living cells. We couple this with single-cell readouts to systematically quantitate the magnitude and heterogeneity of transcriptional responses elicited by each specific chromatin modification. Among these, we show that installing histone H3 lysine 4 trimethylation (H3K4me3) at promoters can causally instruct transcription by hierarchically remodeling the chromatin landscape. We further dissect how DNA sequence motifs influence the transcriptional impact of chromatin marks, identifying switch-like and attenuative effects within distinct cis contexts. Finally, we examine the interplay of combinatorial modifications, revealing that co-targeted H3K27 trimethylation (H3K27me3) and H2AK119 monoubiquitination (H2AK119ub) maximizes silencing penetrance across single cells. Our precision-perturbation strategy unveils the causal principles of how chromatin modification(s) influence transcription and dissects how quantitative responses are calibrated by contextual interactions.
© 2024. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
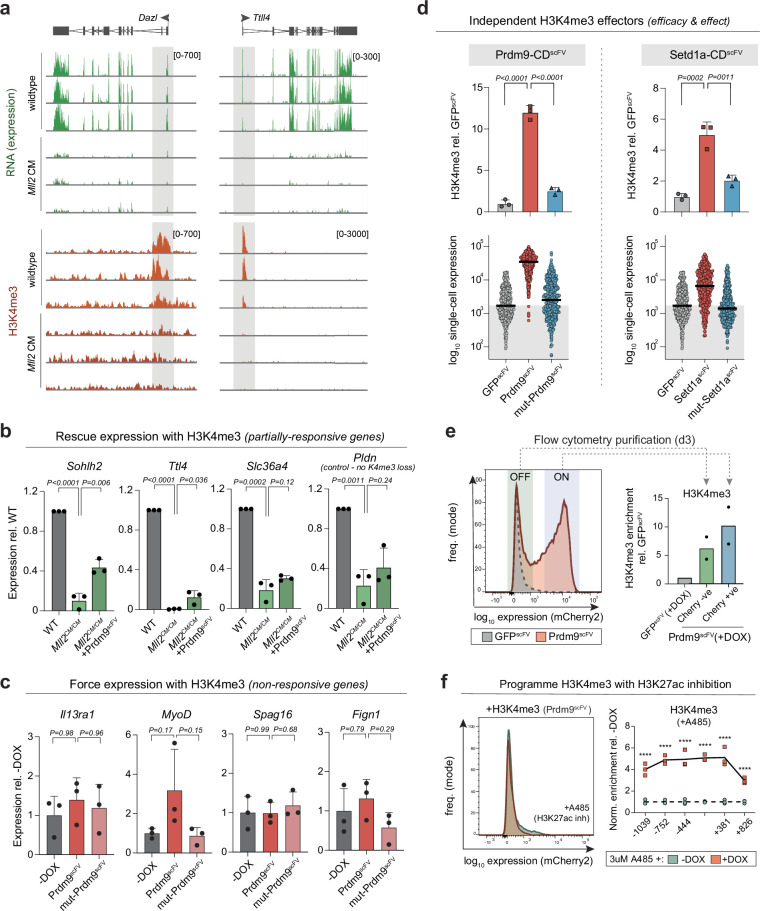
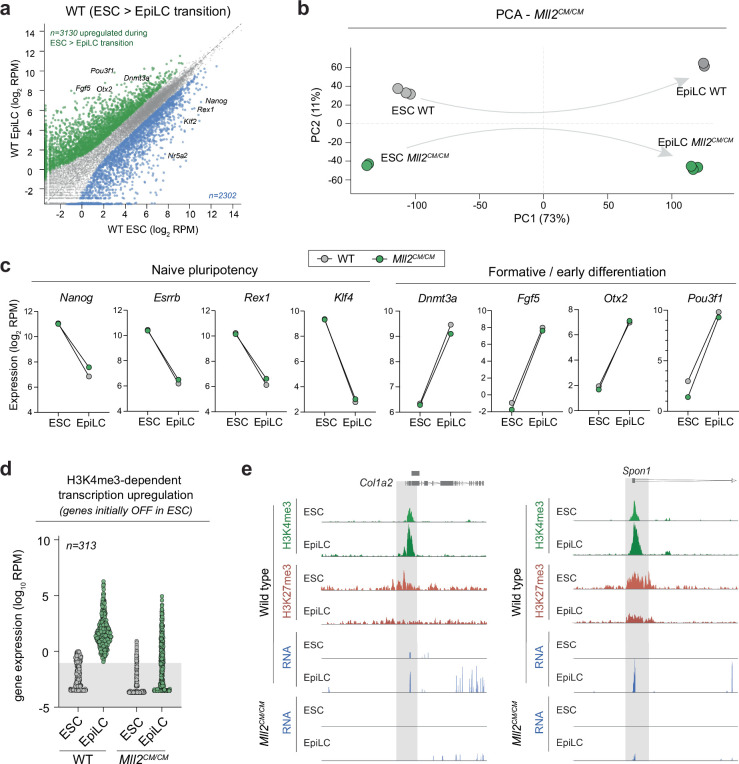
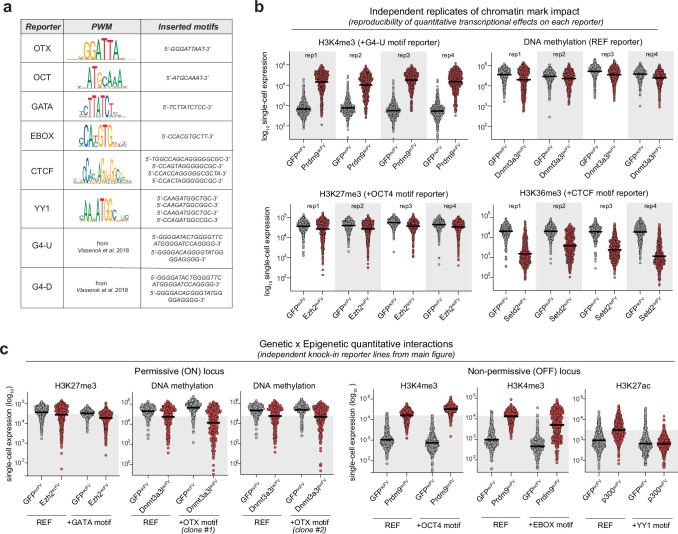
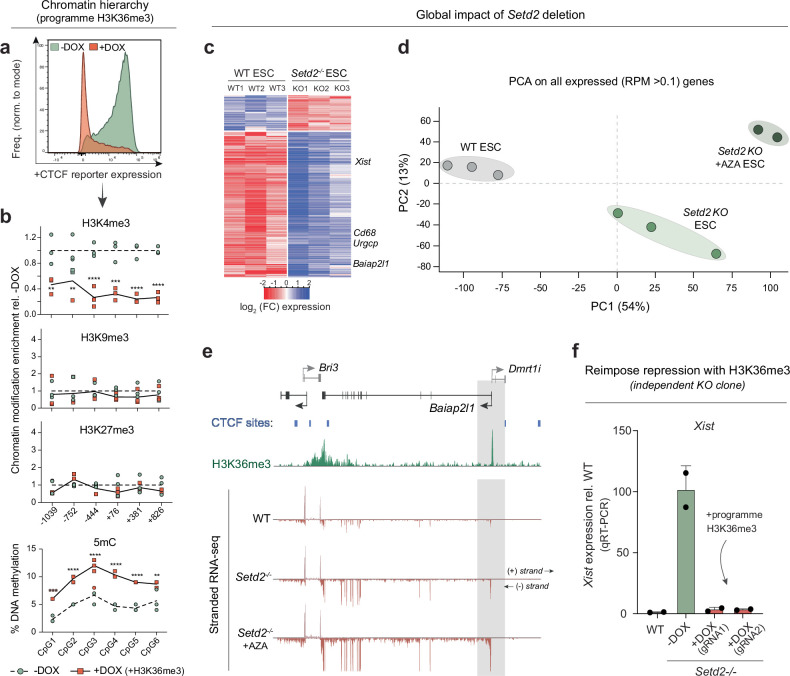
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
