

Increased Expression of α-Hemoglobin Stabilizing Protein (AHSP) mRNA in Erythroid Precursor Cells Isolated from β-Thalassemia Patients Treated with Sirolimus (Rapamycin)
- PMID: 38731008
- PMCID: PMC11084795
- DOI: 10.3390/jcm13092479
Increased Expression of α-Hemoglobin Stabilizing Protein (AHSP) mRNA in Erythroid Precursor Cells Isolated from β-Thalassemia Patients Treated with Sirolimus (Rapamycin)
Abstract
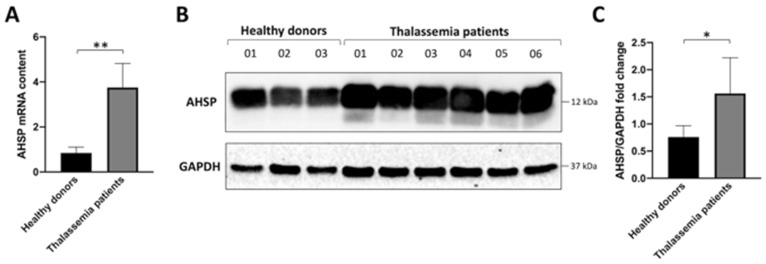
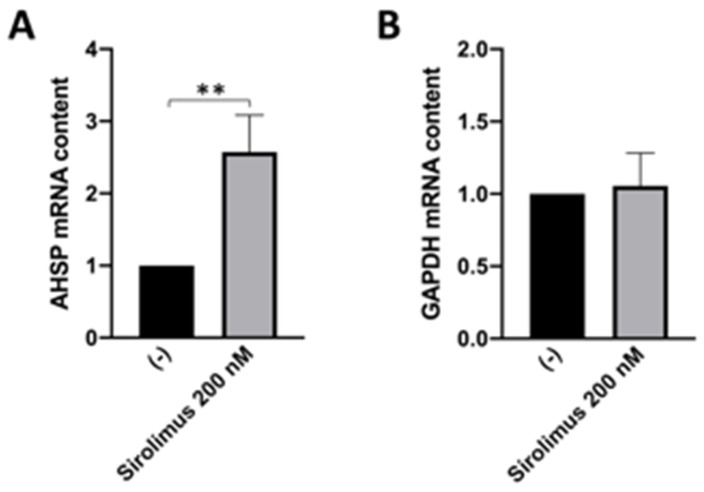
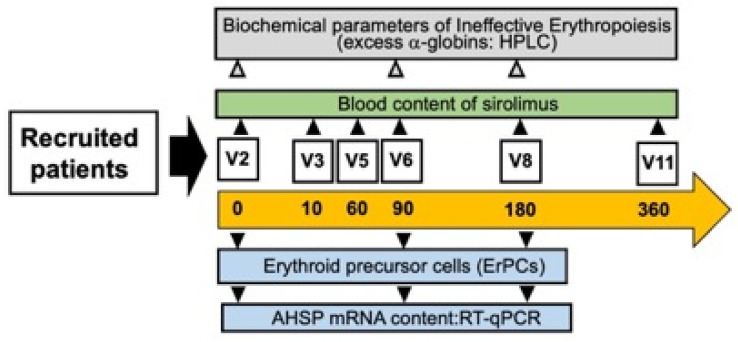
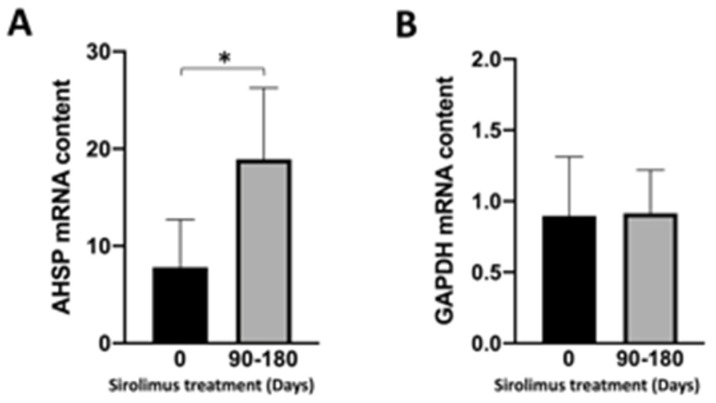
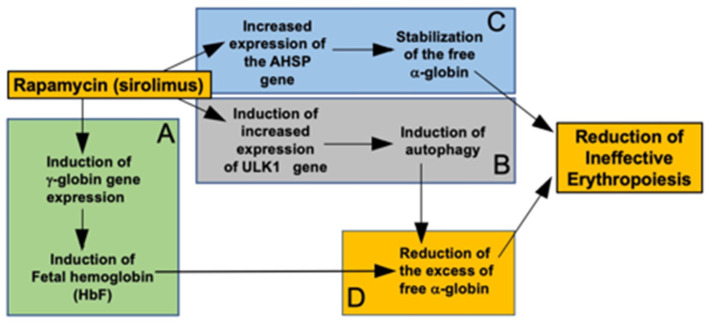
Background/Objectives: in β-thalassemia, important clinical complications are caused by the presence of free α-globin chains in the erythroid cells of β-thalassemia patients. These free α-globin chains are present in excess as a result of the lack of β-globin chains to bind with; they tend to aggregate and precipitate, causing deleterious effects and overall cytotoxicity, maturation arrest of the erythroid cells and, ultimately, ineffective erythropoiesis. The chaperone protein α-hemoglobin-stabilizing protein (AHSP) reversibly binds with free α-globin; the resulting AHSP-αHb complex prevents aggregation and precipitation. Sirolimus (rapamycin) has been previously demonstrated to induce expression of fetal hemoglobin and decrease the excess of free α-globin chain in the erythroid cells of β-thalassemia patients. The objective of this study was to verify whether sirolimus is also able to upregulate AHSP expression in erythroid precursor cells (ErPCs) isolated from β-thalassemia patients. Methods: the expression of AHSP genes was analyzed by measuring the AHSP mRNA content by real-time quantitative PCR (RT-qPCR) and the AHSP protein production by Western blotting. Results: AHSP gene expression was found to be higher in ErPCs of β-thalassemia patients in comparison to ErPCs isolated from healthy subjects. In addition, AHSP expression was further induced by treatment of β-thalassemia ErPCs with sirolimus. Finally, AHSP mRNA was expressed at an increased level in ErPCs of sirolimus-treated β-thalassemia patients participating in the NCT03877809 Sirthalaclin clinical trial. Conclusions: this exploratory study suggests that AHSP expression should be considered as an endpoint in clinical trials based on sirolimus.
Keywords: alpha-hemoglobin stabilizing protein; erythroid precursor cells (ErPCs); rapamycin; sirolimus; β-thalassemia.
Conflict of interest statement
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
Figures
Similar articles
-
Co-Induction of ULK-1 and AHSP mRNAs in Erythroid Precursor Cells Isolated From a Sirolimus-Treated β-Thalassemia Patient: A Case Report Study.Br J Biomed Sci. 2025 Jun 27;82:14311. doi: 10.3389/bjbs.2025.14311. eCollection 2025. Br J Biomed Sci. 2025. PMID: 40655320 Free PMC article.
-
Expression of γ-globin genes in β-thalassemia patients treated with sirolimus: results from a pilot clinical trial (Sirthalaclin).Ther Adv Hematol. 2022 Jun 21;13:20406207221100648. doi: 10.1177/20406207221100648. eCollection 2022. Ther Adv Hematol. 2022. PMID: 35755297 Free PMC article.
-
Decrease in α-Globin and Increase in the Autophagy-Activating Kinase ULK1 mRNA in Erythroid Precursors from β-Thalassemia Patients Treated with Sirolimus.Int J Mol Sci. 2023 Oct 10;24(20):15049. doi: 10.3390/ijms242015049. Int J Mol Sci. 2023. PMID: 37894732 Free PMC article.
-
Alpha-hemoglobin-stabilizing protein (AHSP): a modulatory factor in β-thalassemia.Int J Hematol. 2020 Mar;111(3):352-359. doi: 10.1007/s12185-019-02806-8. Epub 2020 Jan 1. Int J Hematol. 2020. PMID: 31894534 Review.
-
Role of alpha-hemoglobin-stabilizing protein in normal erythropoiesis and beta-thalassemia.Ann N Y Acad Sci. 2005;1054:103-17. doi: 10.1196/annals.1345.013. Ann N Y Acad Sci. 2005. PMID: 16339656 Review.
Cited by
-
Co-Induction of ULK-1 and AHSP mRNAs in Erythroid Precursor Cells Isolated From a Sirolimus-Treated β-Thalassemia Patient: A Case Report Study.Br J Biomed Sci. 2025 Jun 27;82:14311. doi: 10.3389/bjbs.2025.14311. eCollection 2025. Br J Biomed Sci. 2025. PMID: 40655320 Free PMC article.
References
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
