

Distance plus attention for binding affinity prediction
- PMID: 38735985
- PMCID: PMC11089753
- DOI: 10.1186/s13321-024-00844-x
Distance plus attention for binding affinity prediction
Abstract
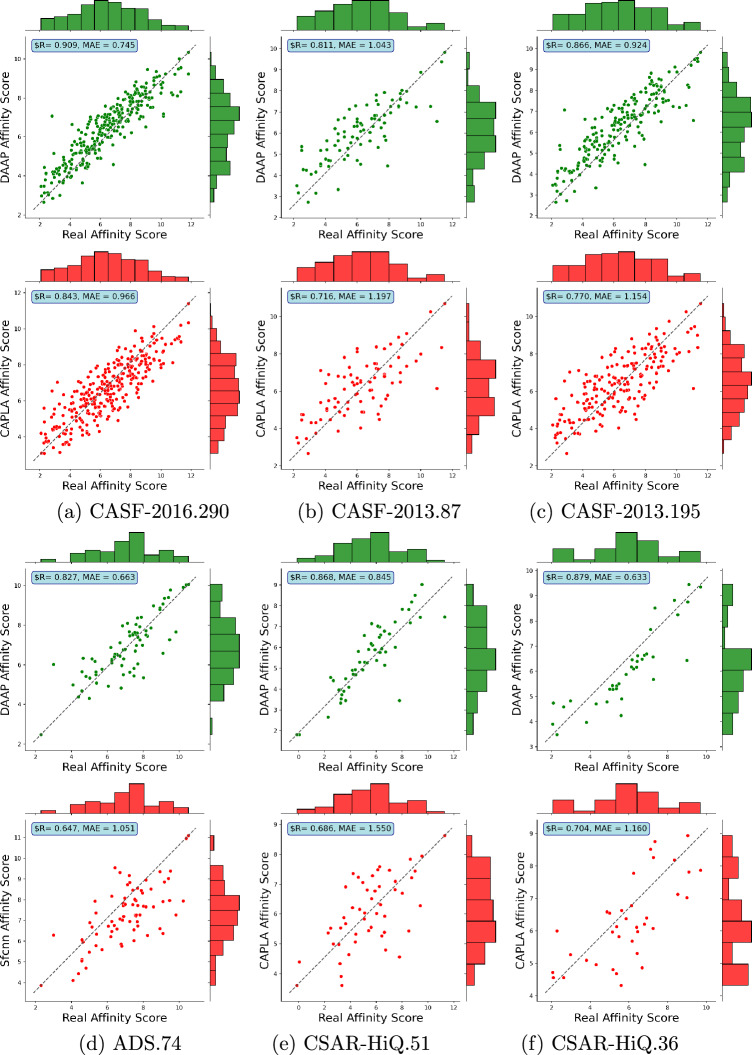
Protein-ligand binding affinity plays a pivotal role in drug development, particularly in identifying potential ligands for target disease-related proteins. Accurate affinity predictions can significantly reduce both the time and cost involved in drug development. However, highly precise affinity prediction remains a research challenge. A key to improve affinity prediction is to capture interactions between proteins and ligands effectively. Existing deep-learning-based computational approaches use 3D grids, 4D tensors, molecular graphs, or proximity-based adjacency matrices, which are either resource-intensive or do not directly represent potential interactions. In this paper, we propose atomic-level distance features and attention mechanisms to capture better specific protein-ligand interactions based on donor-acceptor relations, hydrophobicity, and -stacking atoms. We argue that distances encompass both short-range direct and long-range indirect interaction effects while attention mechanisms capture levels of interaction effects. On the very well-known CASF-2016 dataset, our proposed method, named Distance plus Attention for Affinity Prediction (DAAP), significantly outperforms existing methods by achieving Correlation Coefficient (R) 0.909, Root Mean Squared Error (RMSE) 0.987, Mean Absolute Error (MAE) 0.745, Standard Deviation (SD) 0.988, and Concordance Index (CI) 0.876. The proposed method also shows substantial improvement, around 2% to 37%, on five other benchmark datasets. The program and data are publicly available on the website https://gitlab.com/mahnewton/daap. Scientific Contribution StatementThis study innovatively introduces distance-based features to predict protein-ligand binding affinity, capitalizing on unique molecular interactions. Furthermore, the incorporation of protein sequence features of specific residues enhances the model's proficiency in capturing intricate binding patterns. The predictive capabilities are further strengthened through the use of a deep learning architecture with attention mechanisms, and an ensemble approach, averaging the outputs of five models, is implemented to ensure robust and reliable predictions.
Keywords:
© 2024. The Author(s).
Conflict of interest statement
No Conflict of interest is declared.
Figures





References
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
