

Deep mutational scanning reveals a correlation between degradation and toxicity of thousands of aspartoacylase variants
- PMID: 38740822
- PMCID: PMC11091098
- DOI: 10.1038/s41467-024-48481-0
Deep mutational scanning reveals a correlation between degradation and toxicity of thousands of aspartoacylase variants
Abstract
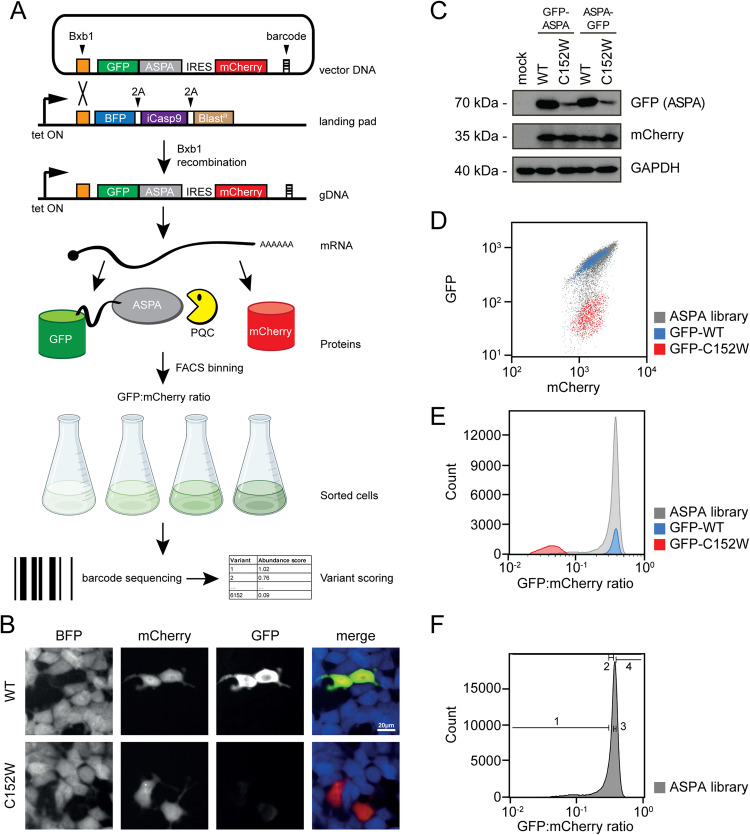
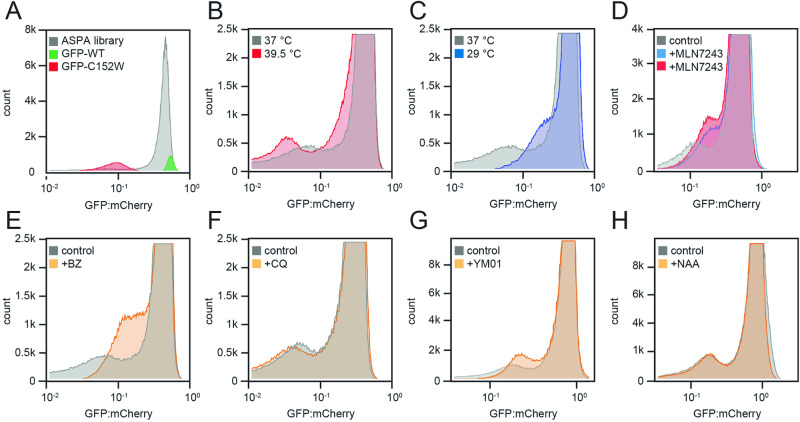
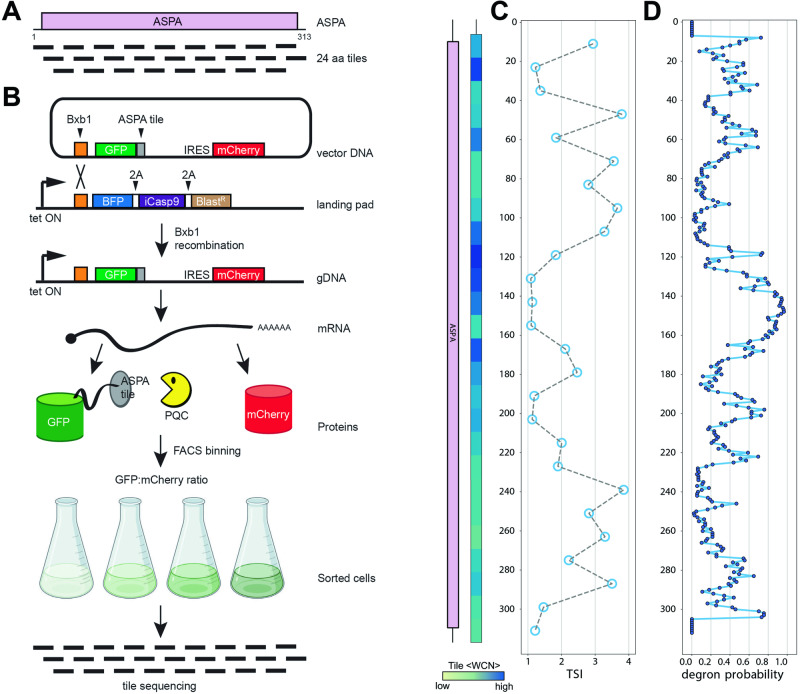
Unstable proteins are prone to form non-native interactions with other proteins and thereby may become toxic. To mitigate this, destabilized proteins are targeted by the protein quality control network. Here we present systematic studies of the cytosolic aspartoacylase, ASPA, where variants are linked to Canavan disease, a lethal neurological disorder. We determine the abundance of 6152 of the 6260 ( ~ 98%) possible single amino acid substitutions and nonsense ASPA variants in human cells. Most low abundance variants are degraded through the ubiquitin-proteasome pathway and become toxic upon prolonged expression. The data correlates with predicted changes in thermodynamic stability, evolutionary conservation, and separate disease-linked variants from benign variants. Mapping of degradation signals (degrons) shows that these are often buried and the C-terminal region functions as a degron. The data can be used to interpret Canavan disease variants and provide insight into the relationship between protein stability, degradation and cell fitness.
© 2024. The Author(s).
Conflict of interest statement
K.L.-L. holds stock options in and is a consultant for Peptone Ltd. The remaining authors declare no competing interests.
Figures




References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
