

Anticancer Drugs of Lysine Specific Histone Demethylase-1 (LSD1) Display Variable Inhibition on Nucleosome Substrates
- PMID: 38742921
- PMCID: PMC12129301
- DOI: 10.1021/acs.biochem.4c00090
Anticancer Drugs of Lysine Specific Histone Demethylase-1 (LSD1) Display Variable Inhibition on Nucleosome Substrates
Abstract
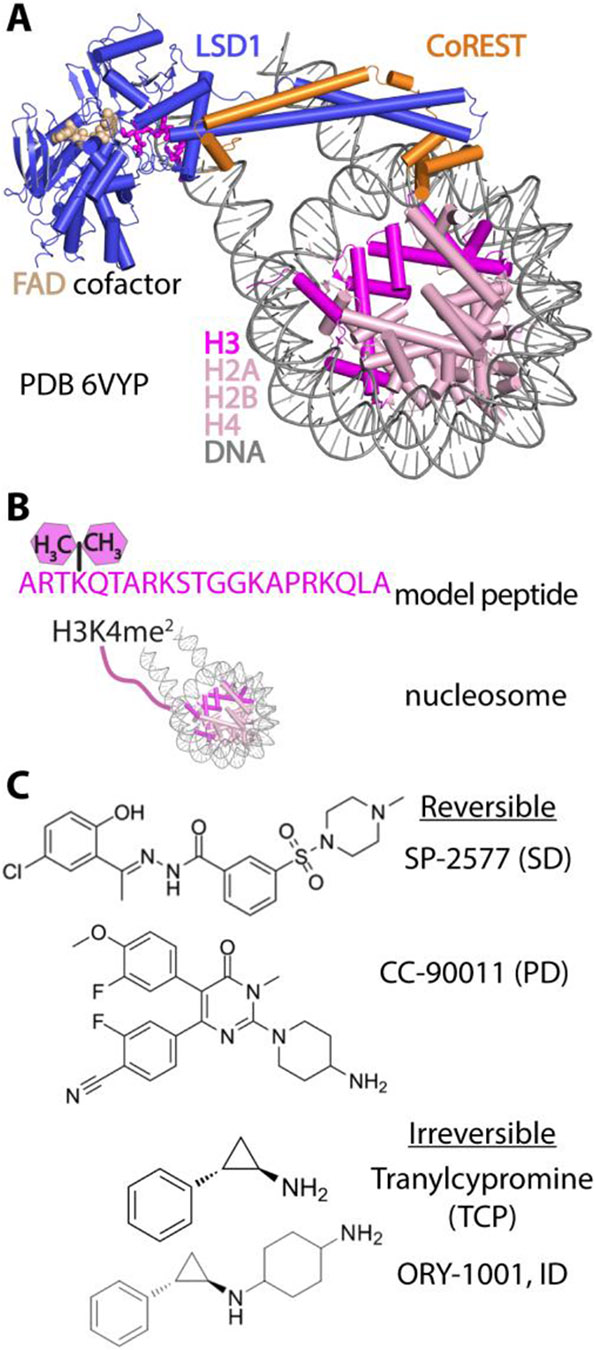
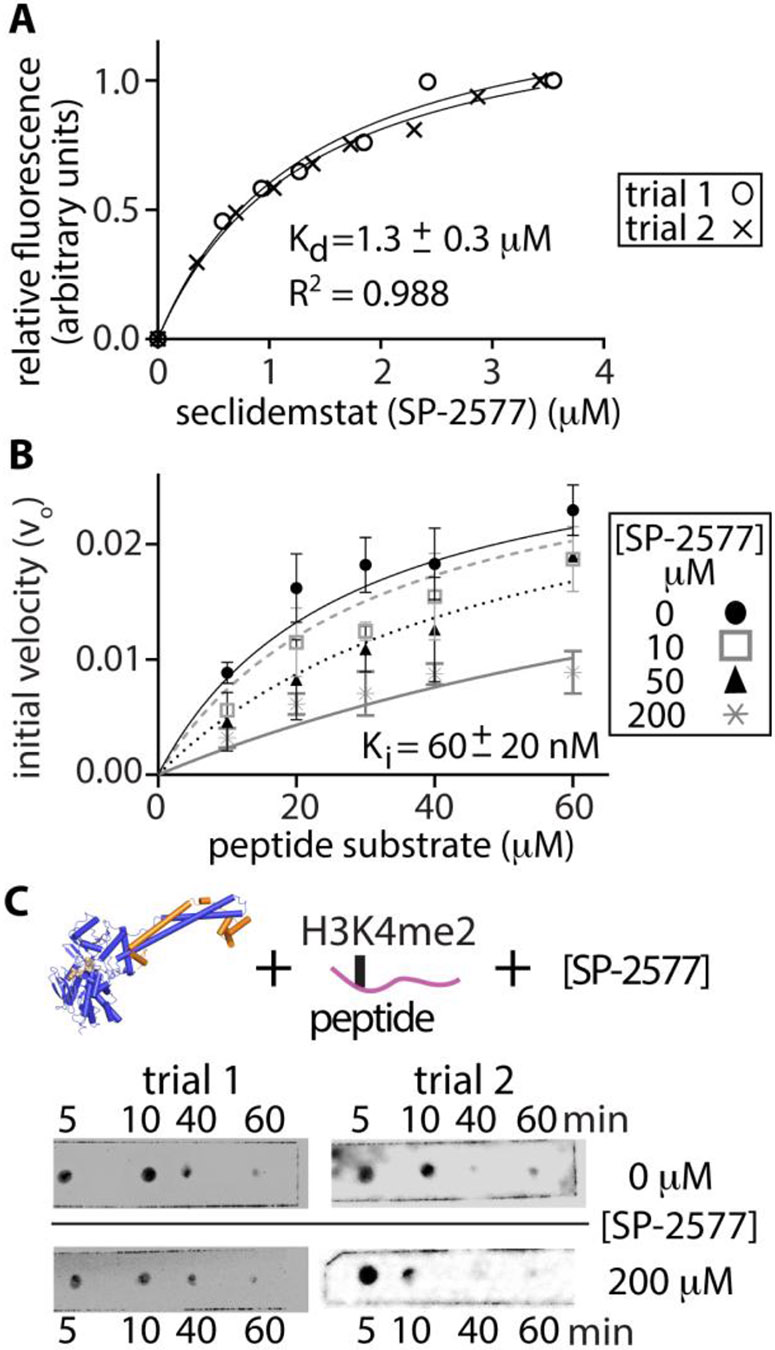
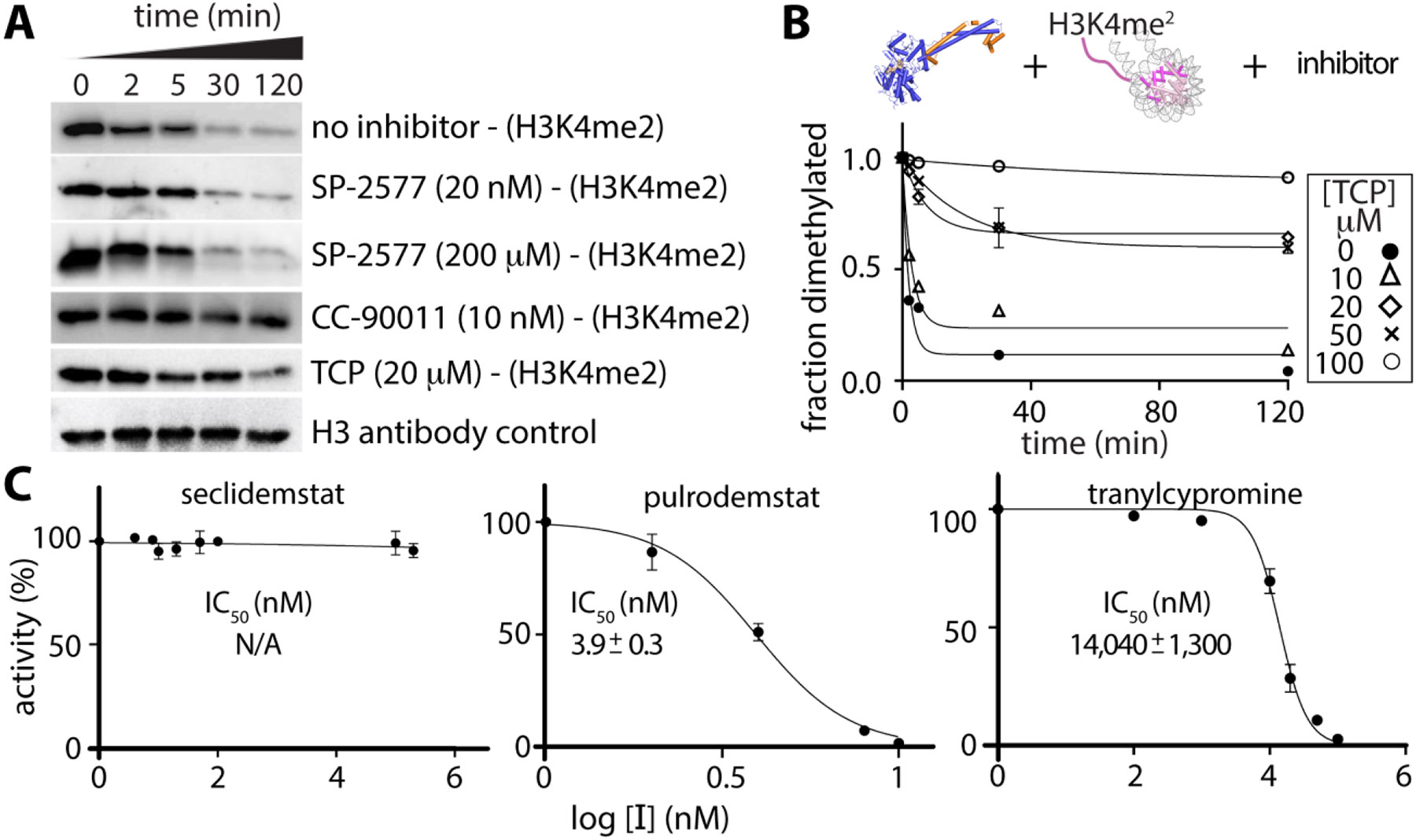
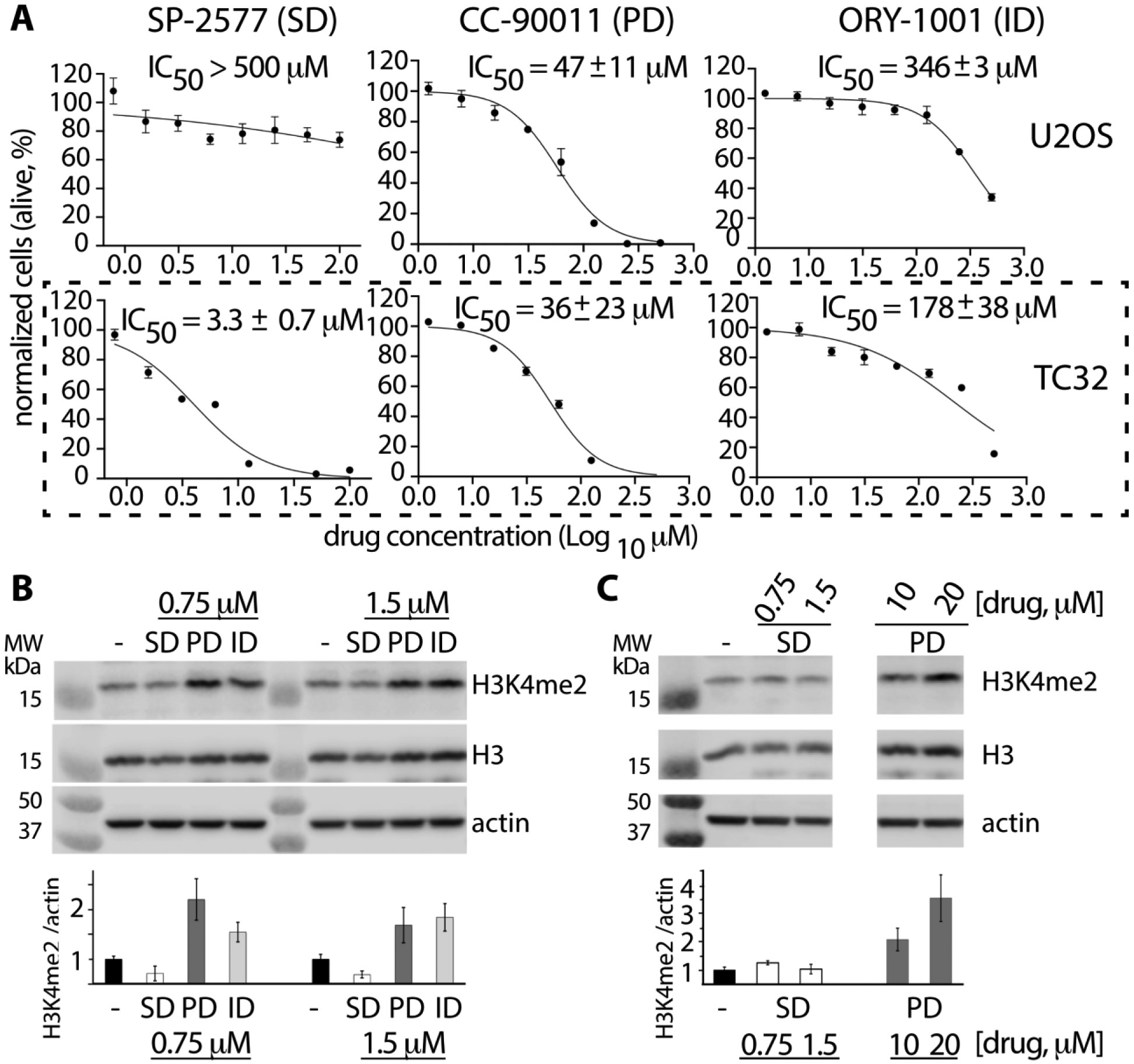
Lysine specific demethylase-1 (LSD1) serves as a regulator of transcription and represents a promising epigenetic target for anticancer treatment. LSD1 inhibitors are in clinical trials for the treatment of Ewing's sarcoma (EWS), acute myeloid leukemia, and small cell lung cancer, and the development of robust inhibitors requires accurate methods for probing demethylation, potency, and selectivity. Here, the inhibition kinetics on the H3K4me2 peptide and nucleosome substrates was examined, comparing the rates of demethylation in the presence of reversible [CC-90011 (PD) and SP-2577 (SD)] and irreversible [ORY-1001 (ID) and tranylcypromine (TCP)] inhibitors. Inhibitors were also subject to viability studies in three human cell lines and Western blot assays to monitor H3K4me2 nucleosome levels in EWS (TC-32) cells, enabling a correlation of drug potency, inhibition in vitro, and cell-based studies. For example, SP-2577, a drug in clinical trials for EWS, inhibits activity on small peptide substrates (Ki = 60 ± 20 nM) using an indirect coupled assay but does not inhibit demethylation on H3K4me2 peptides or nucleosomes using direct Western blot approaches. In addition, the drug has no effect on H3K4me2 levels in TC-32 cells. These data show that SP-2577 is not an LSD1 enzyme inhibitor, although the drug may function independent of demethylation due to its cytotoxic selectivity in TC-32 cells. Taken together, this work highlights the pitfalls of using coupled assays to ascribe a drug's mode of action, emphasizes the use of physiologically relevant substrates in epigenetic drug targeting strategies, and provides insight into the development of substrate-selective inhibitors of LSD1.
Figures
References
-
- Marabelli C; Marrocco B; Mattevi A The growing structural and functional complexity of the LSD1/KDM1A histone demethylase. Curr. Opin Struct Biol 2016, 41, 135–144. - PubMed
-
- Shi Y; Lan F; Matson C; Mulligan P; Whetstine JR; Cole PA; Casero RA; Shi Y Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell 2004, 119, 941–953. - PubMed
-
- Yang M; Gocke CB; Luo X; Borek D; Tomchick DR; Machius M; Otwinowski Z; Yu H Structural basis for CoREST-dependent demethylation of nucleosomes by the human LSD1histone demethylase. Mol. Cell 2006, 23, 377–387. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
