

Analyzing the functional effects of DNA variants with gene editing
- PMID: 38744287
- PMCID: PMC11133854
- DOI: 10.1016/j.crmeth.2024.100776
Analyzing the functional effects of DNA variants with gene editing
Abstract
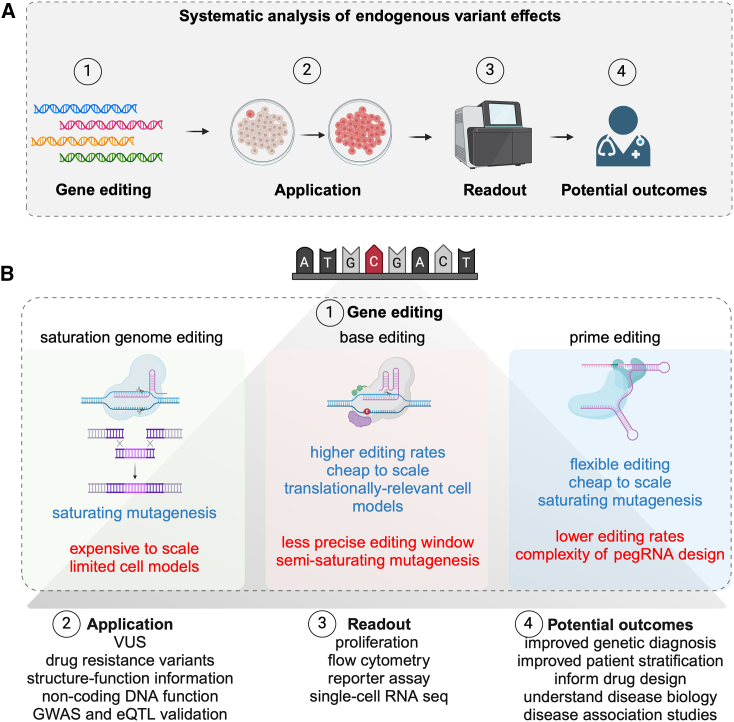
Continual advancements in genomics have led to an ever-widening disparity between the rate of discovery of genetic variants and our current understanding of their functions and potential roles in disease. Systematic methods for phenotyping DNA variants are required to effectively translate genomics data into improved outcomes for patients with genetic diseases. To make the biggest impact, these approaches must be scalable and accurate, faithfully reflect disease biology, and define complex disease mechanisms. We compare current methods to analyze the function of variants in their endogenous DNA context using genome editing strategies, such as saturation genome editing, base editing and prime editing. We discuss how these technologies can be linked to high-content readouts to gain deep mechanistic insights into variant effects. Finally, we highlight key challenges that need to be addressed to bridge the genotype to phenotype gap, and ultimately improve the diagnosis and treatment of genetic diseases.
Keywords: CP: biotechnology; CP: genetics.
Copyright © 2024 The Author(s). Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests A.R.B. has been a founder and consultant for EnsoCell since August 2023.
Figures


References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
