

Genetics of inherited peripheral neuropathies and the next frontier: looking backwards to progress forwards
- PMID: 38744462
- PMCID: PMC11503175
- DOI: 10.1136/jnnp-2024-333436
Genetics of inherited peripheral neuropathies and the next frontier: looking backwards to progress forwards
Abstract
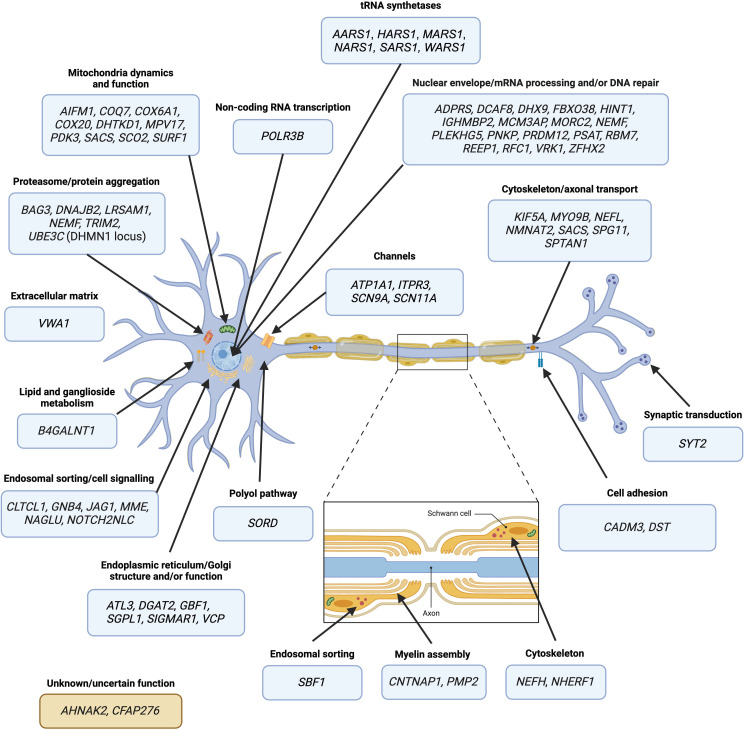
Inherited peripheral neuropathies (IPNs) encompass a clinically and genetically heterogeneous group of disorders causing length-dependent degeneration of peripheral autonomic, motor and/or sensory nerves. Despite gold-standard diagnostic testing for pathogenic variants in over 100 known associated genes, many patients with IPN remain genetically unsolved. Providing patients with a diagnosis is critical for reducing their 'diagnostic odyssey', improving clinical care, and for informed genetic counselling. The last decade of massively parallel sequencing technologies has seen a rapid increase in the number of newly described IPN-associated gene variants contributing to IPN pathogenesis. However, the scarcity of additional families and functional data supporting variants in potential novel genes is prolonging patient diagnostic uncertainty and contributing to the missing heritability of IPNs. We review the last decade of IPN disease gene discovery to highlight novel genes, structural variation and short tandem repeat expansions contributing to IPN pathogenesis. From the lessons learnt, we provide our vision for IPN research as we anticipate the future, providing examples of emerging technologies, resources and tools that we propose that will expedite the genetic diagnosis of unsolved IPN families.
Keywords: GENETICS; HMSN (CHARCOT-MARIE-TOOTH); NEUROGENETICS; NEUROMUSCULAR; NEUROPATHY.
© Author(s) (or their employer(s)) 2024. Re-use permitted under CC BY. Published by BMJ.
Conflict of interest statement
Competing interests: None declared.
Figures
Similar articles
-
Structural variations causing inherited peripheral neuropathies: A paradigm for understanding genomic organization, chromatin interactions, and gene dysregulation.Mol Genet Genomic Med. 2018 May;6(3):422-433. doi: 10.1002/mgg3.390. Epub 2018 Mar 23. Mol Genet Genomic Med. 2018. PMID: 29573232 Free PMC article. Review.
-
Whole-exome sequencing is a valuable diagnostic tool for inherited peripheral neuropathies: Outcomes from a cohort of 50 families.Clin Genet. 2018 Feb;93(2):301-309. doi: 10.1111/cge.13101. Epub 2017 Dec 12. Clin Genet. 2018. PMID: 28708278
-
Identification of Genetic Causes of Inherited Peripheral Neuropathies by Targeted Gene Panel Sequencing.Mol Cells. 2016 May 31;39(5):382-8. doi: 10.14348/molcells.2016.2288. Epub 2016 Mar 30. Mol Cells. 2016. PMID: 27025386 Free PMC article.
-
Linking LRP12 CGG repeat expansion to inherited peripheral neuropathy.J Neurol Neurosurg Psychiatry. 2025 Jan 16;96(2):140-149. doi: 10.1136/jnnp-2024-333403. J Neurol Neurosurg Psychiatry. 2025. PMID: 39013564 Free PMC article.
-
Molecular pathogenesis of peripheral neuropathies: insights from Drosophila models.Curr Opin Genet Dev. 2017 Jun;44:61-73. doi: 10.1016/j.gde.2017.01.011. Epub 2017 Feb 16. Curr Opin Genet Dev. 2017. PMID: 28213160 Review.
Cited by
-
Nationwide Phenotypic and Genotypic Characterisation of 103 Patients With SH3TC2 Gene-Related Demyelinating Peripheral Neuropathy.Eur J Neurol. 2025 Aug;32(8):e70313. doi: 10.1111/ene.70313. Eur J Neurol. 2025. PMID: 40745932 Free PMC article.
-
Clinical and Genetic Reassessment in Patients With Clinically Diagnosed Hereditary Polyneuropathy.Eur J Neurol. 2025 Aug;32(8):e70301. doi: 10.1111/ene.70301. Eur J Neurol. 2025. PMID: 40755160 Free PMC article.
-
Molecular diagnostic approach to rare neurological diseases from a clinician viewpoint.Genomics Inform. 2024 Oct 10;22(1):18. doi: 10.1186/s44342-024-00025-0. Genomics Inform. 2024. PMID: 39390516 Free PMC article. Review.
-
Expanding the genetic spectrum of hereditary motor sensory neuropathies in Pakistan.BMC Neurol. 2024 Oct 16;24(1):394. doi: 10.1186/s12883-024-03882-y. BMC Neurol. 2024. PMID: 39415096 Free PMC article.
-
A systematic review of hereditary neurological disorders diagnosed by whole exome sequencing in Pakistani population: updates from 2014 to November 2024.Neurogenetics. 2025 Apr 3;26(1):40. doi: 10.1007/s10048-025-00819-6. Neurogenetics. 2025. PMID: 40178666
References
Publication types
MeSH terms
Supplementary concepts
LinkOut - more resources
Full Text Sources
Medical