

Semiconservative transmission of DNA N 6-adenine methylation in a unicellular eukaryote
- PMID: 38744529
- PMCID: PMC11216311
- DOI: 10.1101/gr.277843.123
Semiconservative transmission of DNA N 6-adenine methylation in a unicellular eukaryote
Abstract
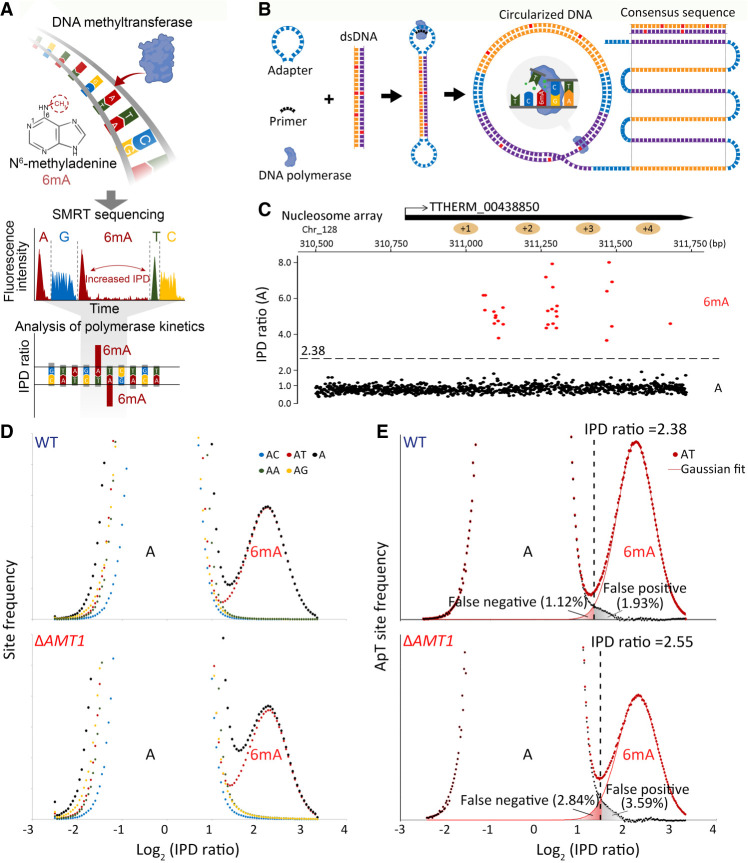
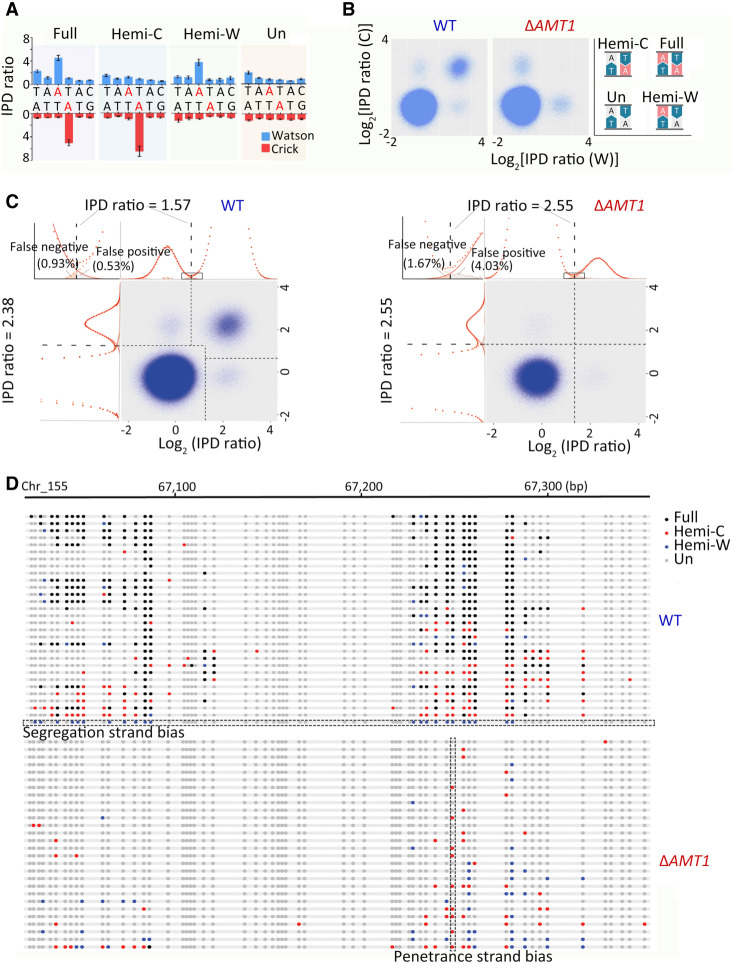
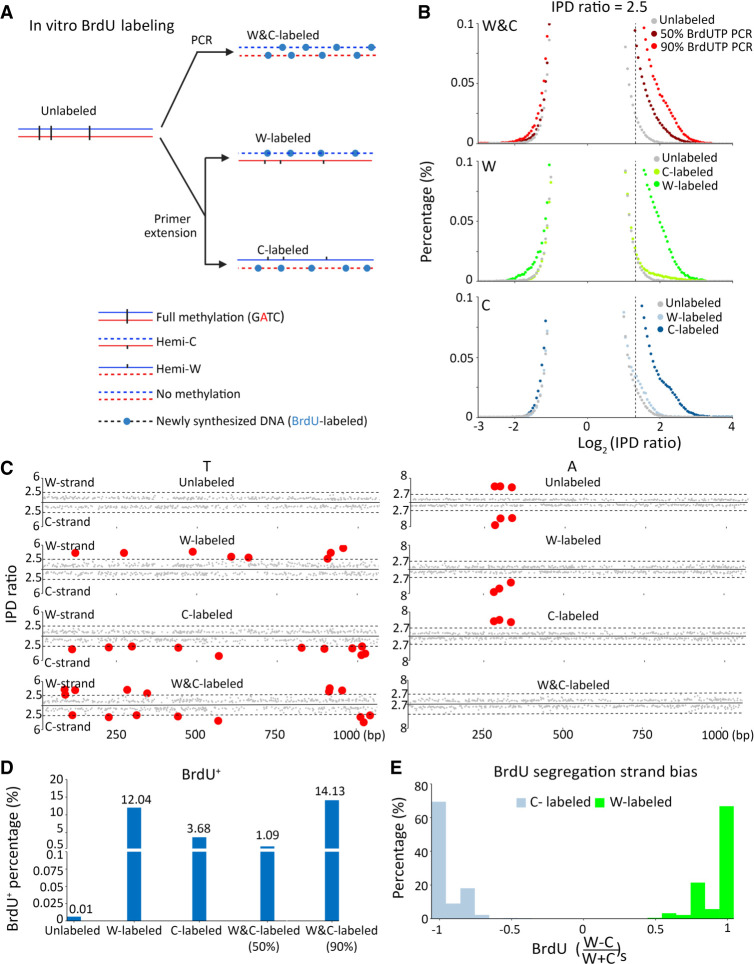
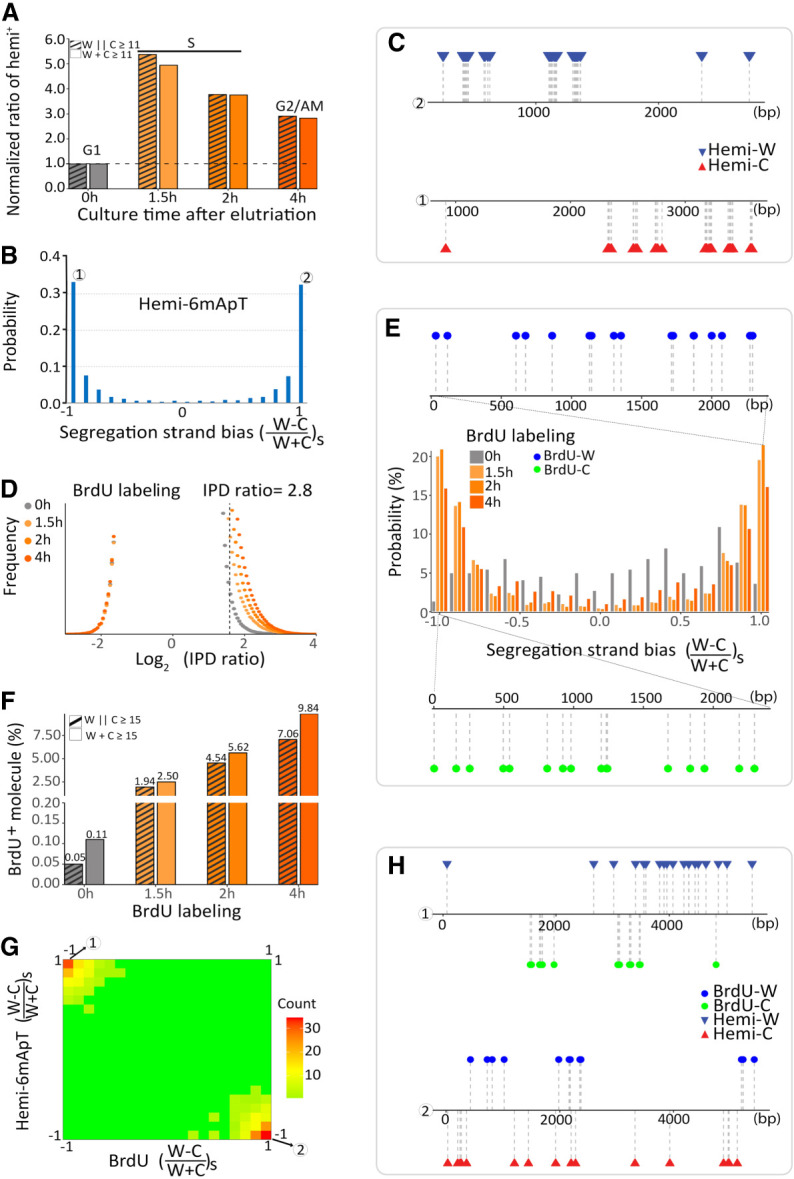
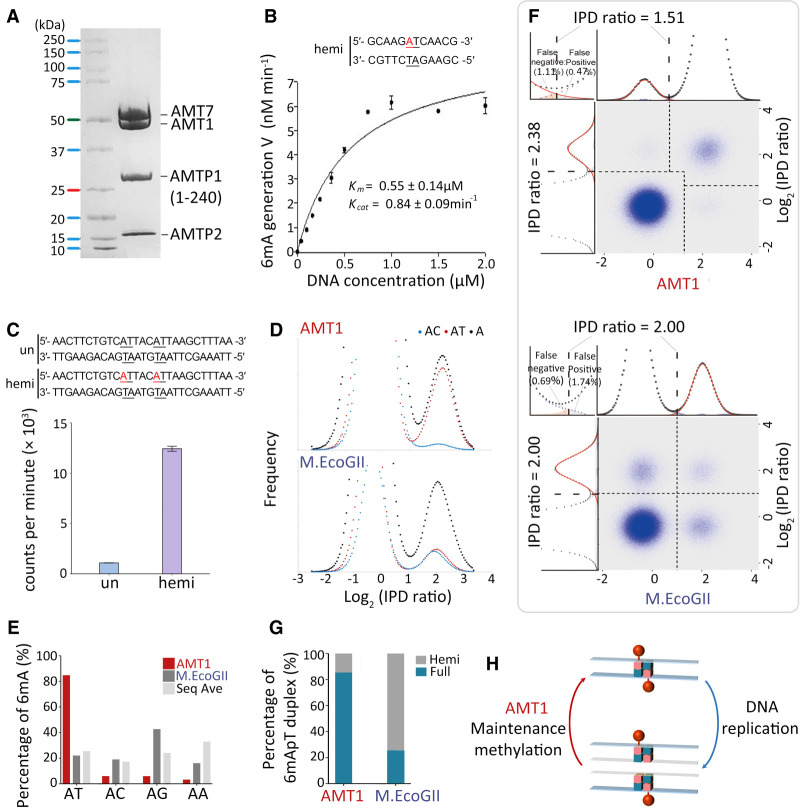
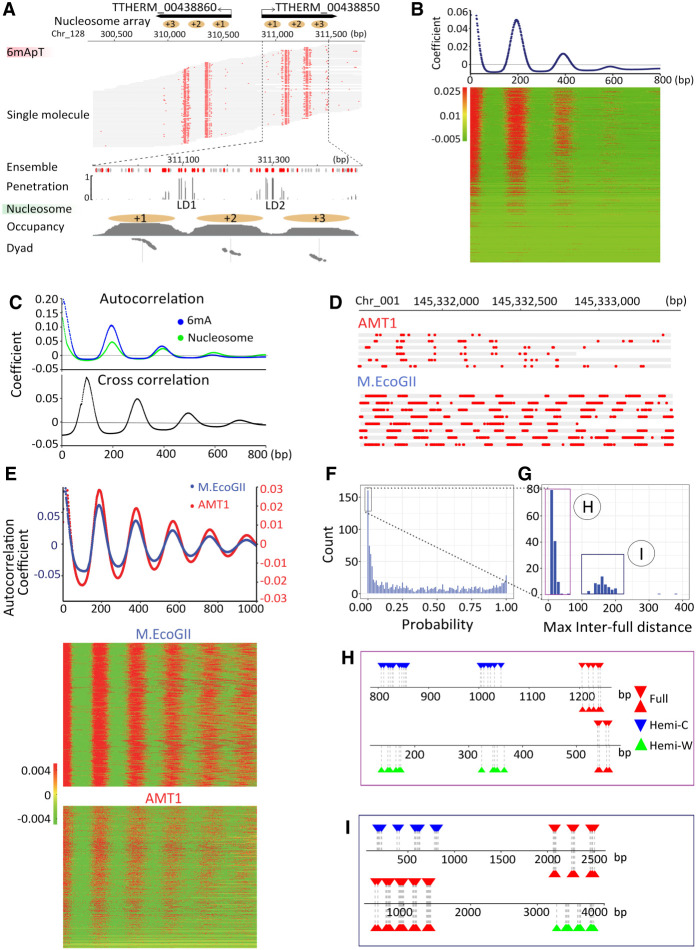
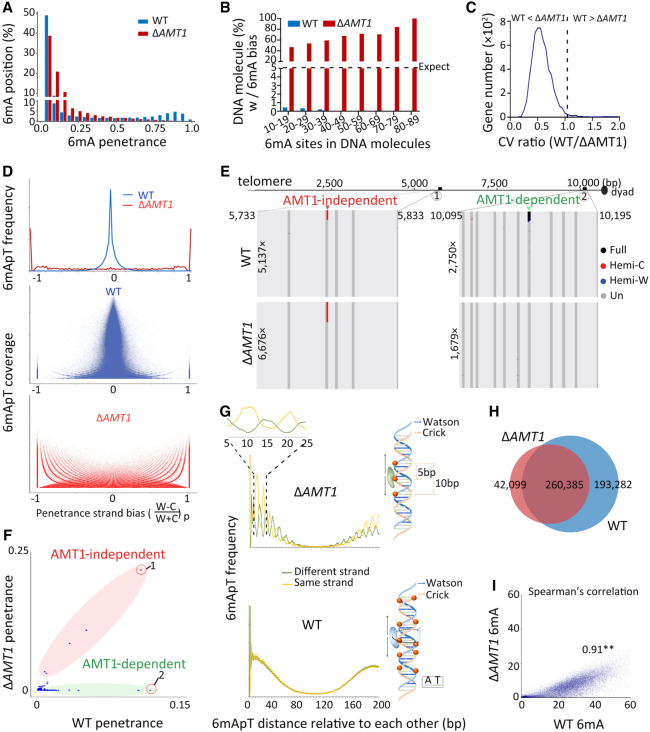
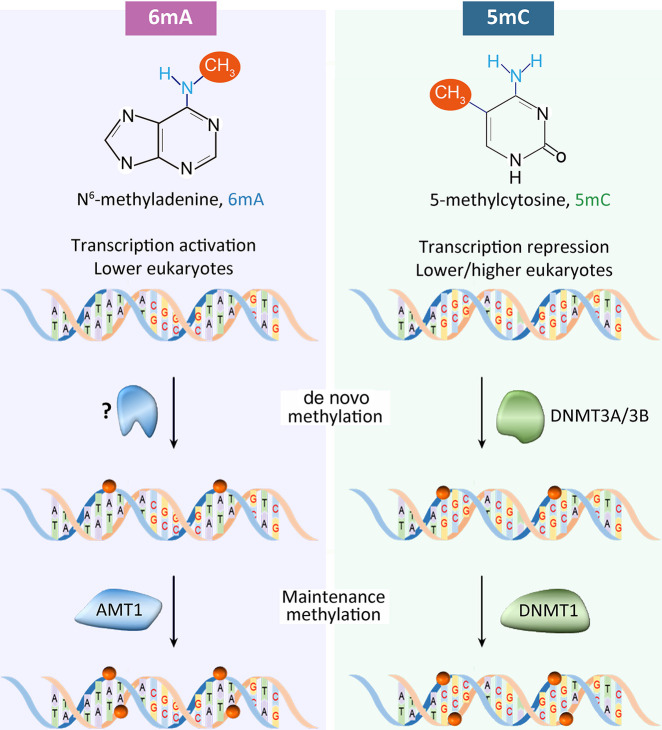
Although DNA N 6-adenine methylation (6mA) is best known in prokaryotes, its presence in eukaryotes has recently generated great interest. Biochemical and genetic evidence supports that AMT1, an MT-A70 family methyltransferase (MTase), is crucial for 6mA deposition in unicellular eukaryotes. Nonetheless, the 6mA transmission mechanism remains to be elucidated. Taking advantage of single-molecule real-time circular consensus sequencing (SMRT CCS), here we provide definitive evidence for semiconservative transmission of 6mA in Tetrahymena thermophila In wild-type (WT) cells, 6mA occurs at the self-complementary ApT dinucleotide, mostly in full methylation (full-6mApT); after DNA replication, hemi-methylation (hemi-6mApT) is transiently present on the parental strand, opposite to the daughter strand readily labeled by 5-bromo-2'-deoxyuridine (BrdU). In ΔAMT1 cells, 6mA predominantly occurs as hemi-6mApT. Hemi-to-full conversion in WT cells is fast, robust, and processive, whereas de novo methylation in ΔAMT1 cells is slow and sporadic. In Tetrahymena, regularly spaced 6mA clusters coincide with the linker DNA of nucleosomes arrayed in the gene body. Importantly, in vitro methylation of human chromatin by the reconstituted AMT1 complex recapitulates preferential targeting of hemi-6mApT sites in linker DNA, supporting AMT1's intrinsic and autonomous role in maintenance methylation. We conclude that 6mA is transmitted by a semiconservative mechanism: full-6mApT is split by DNA replication into hemi-6mApT, which is restored to full-6mApT by AMT1-dependent maintenance methylation. Our study dissects AMT1-dependent maintenance methylation and AMT1-independent de novo methylation, reveals a 6mA transmission pathway with a striking similarity to 5-methylcytosine (5mC) transmission at the CpG dinucleotide, and establishes 6mA as a bona fide eukaryotic epigenetic mark.
© 2024 Sheng et al.; Published by Cold Spring Harbor Laboratory Press.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources