

Systematic dissection of tumor-normal single-cell ecosystems across a thousand tumors of 30 cancer types
- PMID: 38744958
- PMCID: PMC11094150
- DOI: 10.1038/s41467-024-48310-4
Systematic dissection of tumor-normal single-cell ecosystems across a thousand tumors of 30 cancer types
Erratum in
-
Author Correction: Systematic dissection of tumor-normal single-cell ecosystems across a thousand tumors of 30 cancer types.Nat Commun. 2025 Mar 21;16(1):2806. doi: 10.1038/s41467-025-58068-y. Nat Commun. 2025. PMID: 40118866 Free PMC article. No abstract available.
Abstract
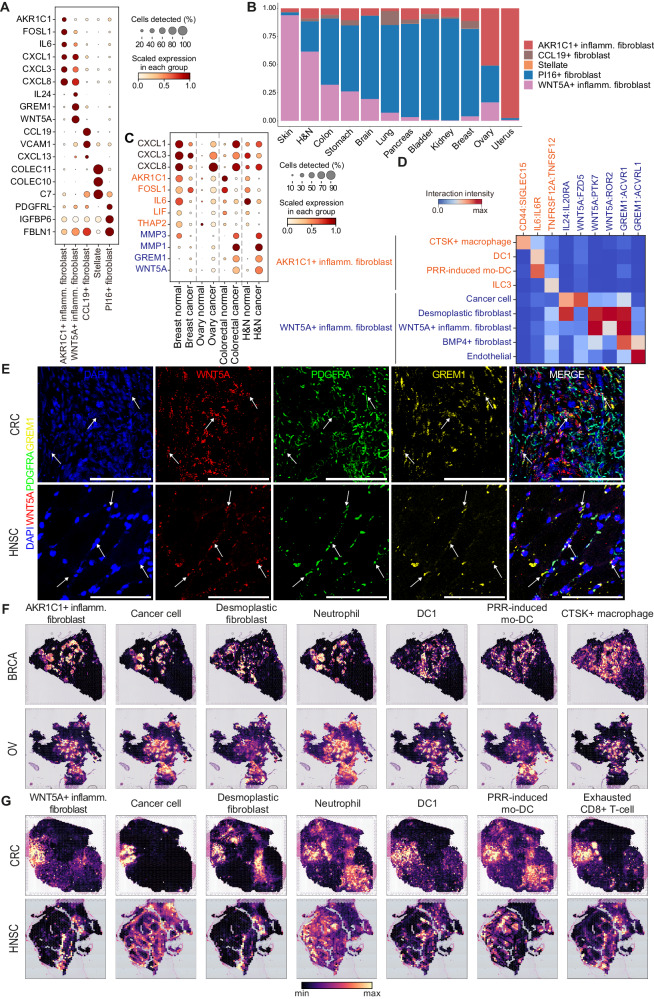
The complexity of the tumor microenvironment poses significant challenges in cancer therapy. Here, to comprehensively investigate the tumor-normal ecosystems, we perform an integrative analysis of 4.9 million single-cell transcriptomes from 1070 tumor and 493 normal samples in combination with pan-cancer 137 spatial transcriptomics, 8887 TCGA, and 1261 checkpoint inhibitor-treated bulk tumors. We define a myriad of cell states constituting the tumor-normal ecosystems and also identify hallmark gene signatures across different cell types and organs. Our atlas characterizes distinctions between inflammatory fibroblasts marked by AKR1C1 or WNT5A in terms of cellular interactions and spatial co-localization patterns. Co-occurrence analysis reveals interferon-enriched community states including tertiary lymphoid structure (TLS) components, which exhibit differential rewiring between tumor, adjacent normal, and healthy normal tissues. The favorable response of interferon-enriched community states to immunotherapy is validated using immunotherapy-treated cancers (n = 1261) including our lung cancer cohort (n = 497). Deconvolution of spatial transcriptomes discriminates TLS-enriched from non-enriched cell types among immunotherapy-favorable components. Our systematic dissection of tumor-normal ecosystems provides a deeper understanding of inter- and intra-tumoral heterogeneity.
© 2024. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures





References
-
- Hanahan, D. & Weinberg, R. A. Hallmarks of cancer: the next generation. Cell144, 646–674 (2011). - PubMed
-
- Bassez, A. et al. A single-cell map of intratumoral changes during anti-PD1 treatment of patients with breast cancer. Nat. Med.27, 820–832 (2021). - PubMed
-
- Cheng, S. et al. A pan-cancer single-cell transcriptional atlas of tumor infiltrating myeloid cells. Cell184, 792–809.e23 (2021). - PubMed
MeSH terms
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
