

This is a preprint.
Addressing pandemic-wide systematic errors in the SARS-CoV-2 phylogeny
- PMID: 38746185
- PMCID: PMC11092452
- DOI: 10.1101/2024.04.29.591666
Addressing pandemic-wide systematic errors in the SARS-CoV-2 phylogeny
Update in
-
Addressing pandemic-wide systematic errors in the SARS-CoV-2 phylogeny.Nat Methods. 2026 Mar;23(3):653-662. doi: 10.1038/s41592-025-02947-1. Epub 2026 Feb 9. Nat Methods. 2026. PMID: 41663577 Free PMC article.
Abstract
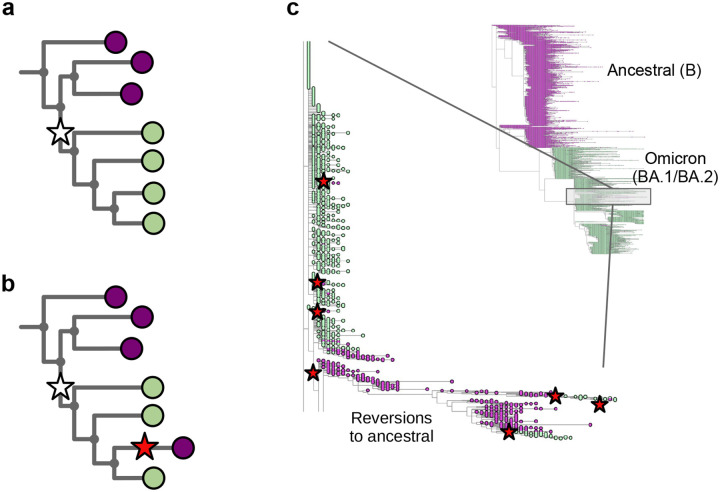
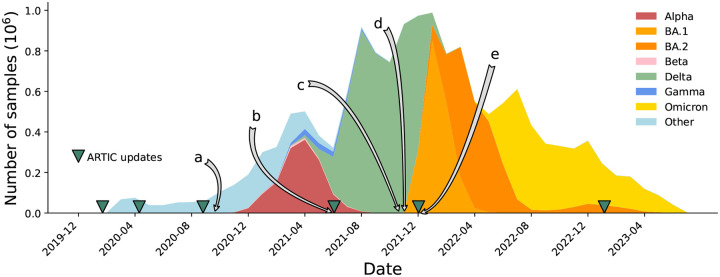
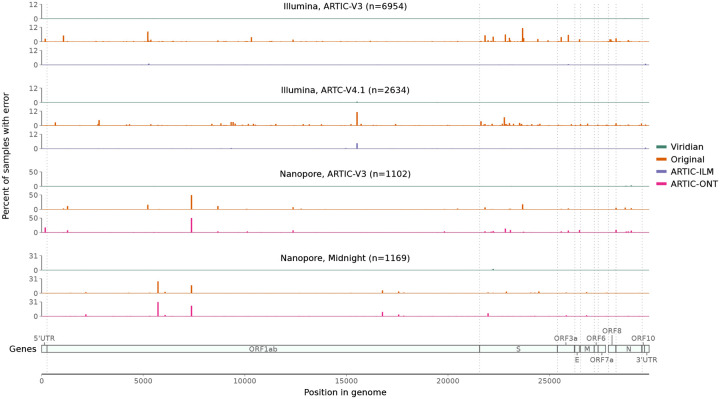
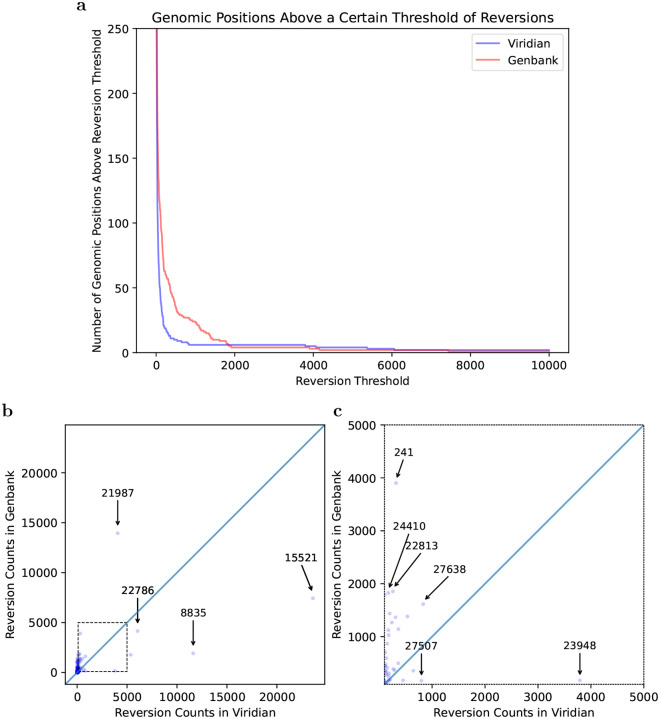
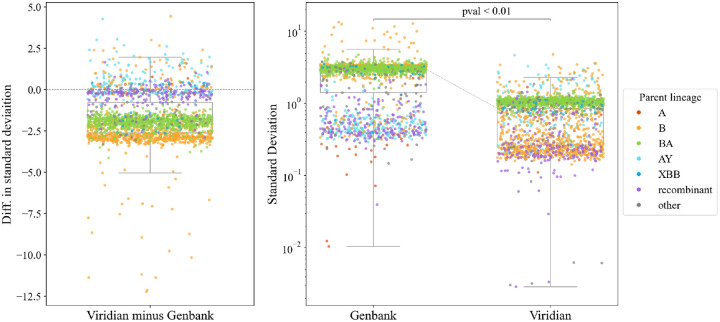
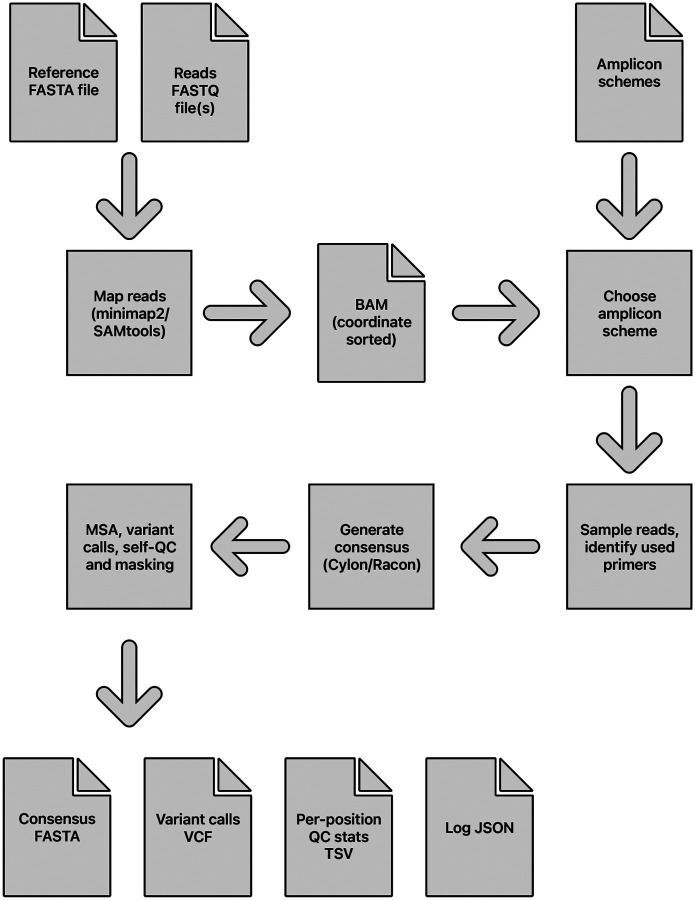
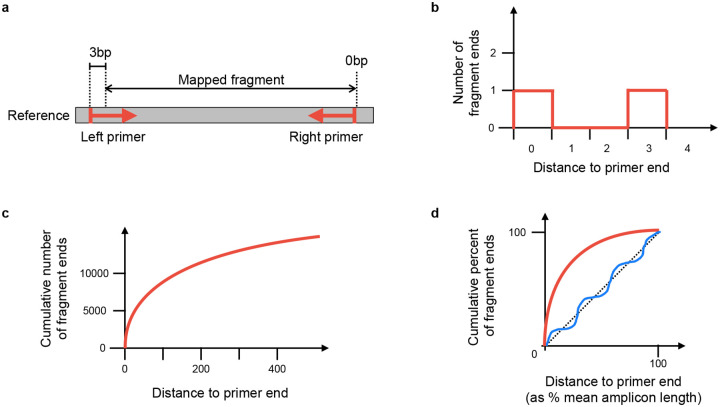
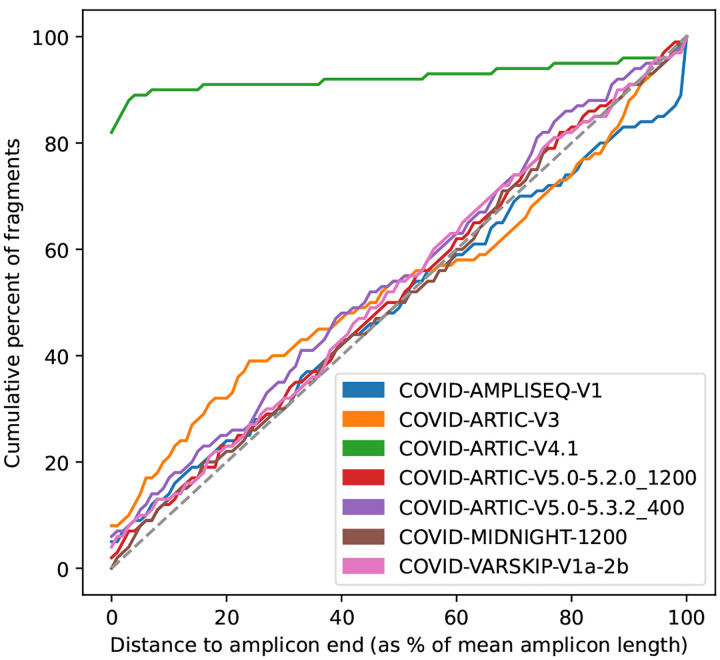
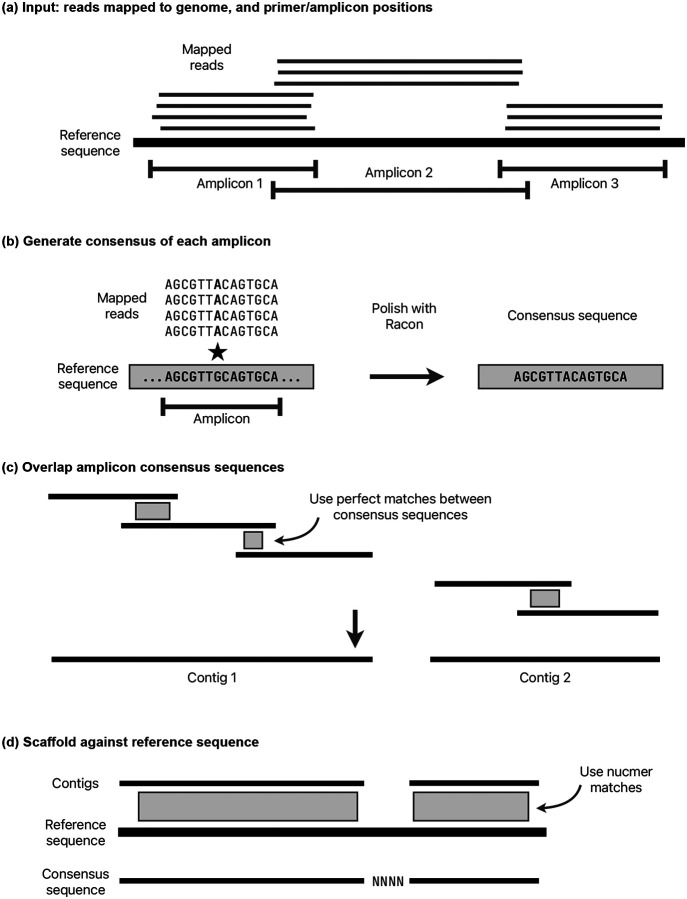
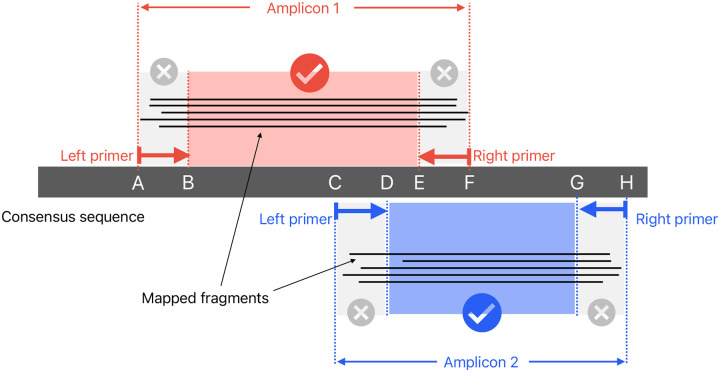
The SARS-CoV-2 genome occupies a unique place in infection biology - it is the most highly sequenced genome on earth (making up over 20% of public sequencing datasets) with fine scale information on sampling date and geography, and has been subject to unprecedented intense analysis. As a result, these phylogenetic data are an incredibly valuable resource for science and public health. However, the vast majority of the data was sequenced by tiling amplicons across the full genome, with amplicon schemes that changed over the pandemic as mutations in the viral genome interacted with primer binding sites. In combination with the disparate set of genome assembly workflows and lack of consistent quality control (QC) processes, the current genomes have many systematic errors that have evolved with the virus and amplicon schemes. These errors have significant impacts on the phylogeny, and therefore over the last few years, many thousands of hours of researchers time has been spent in "eyeballing" trees, looking for artefacts, and then patching the tree. Given the huge value of this dataset, we therefore set out to reprocess the complete set of public raw sequence data in a rigorous amplicon-aware manner, and build a cleaner phylogeny. Here we provide a global tree of 4,471,579 samples, built from a consistently assembled set of high quality consensus sequences from all available public data as of June 2024, viewable at https://viridian.taxonium.org. Each genome was constructed using a novel assembly tool called Viridian (https://github.com/iqbal-lab-org/viridian), developed specifically to process amplicon sequence data, eliminating artefactual errors and mask the genome at low quality positions. We provide simulation and empirical validation of the methodology, and quantify the improvement in the phylogeny. We hope the tree, consensus sequences and Viridian will be a valuable resource for researchers.
Conflict of interest statement
Conflict of Interest Gavin Screaton sits on the GSK Vaccines Scientific Advisory Board, consults for AstraZeneca, and is a founding member of RQ Biotechnology.
Figures
References
-
- Turakhia Yatish, De Maio Nicola, Thornlow Bryan, Gozashti Landen, Lanfear Robert, Walker Conor R., Hinrichs Angie S., Fernandes Jason D., Borges Rui, Slodkowicz Greg, Weilguny Lukas, Haussler David, Goldman Nick, and Russell Corbett-Detig. Stability of SARS-CoV-2 phylogenies. PLOS Genetics, 16(11):e1009175, November 2020. - PMC - PubMed
-
- De Maio Nicola, Walker Conor, Borges Rui, Weilguny Lukas, Slodkowicz Greg, and Goldman Nick. Issues with sars-cov-2 sequencing data, https://virological.org/t/issues-with-sars-cov-2-sequencing-data/473. May 2020.
-
- Henn Matthew R., Boutwell Christian L., Charlebois Patrick, Lennon Niall J., Power Karen A., Macalalad Alexander R., Berlin Aaron M., Malboeuf Christine M., Ryan Elizabeth M., Gnerre Sante, Zody Michael C., Erlich Rachel L., Green Lisa M., Berical Andrew, Wang Yaoyu, Casali Monica, Streeck Hendrik, Bloom Allyson K., Dudek Tim, Tully Damien, Newman Ruchi, Axten Karen L., Gladden Adrianne D., Battis Laura, Kemper Michael, Zeng Qiandong, Shea Terrance P., Gujja Sharvari, Zedlack Carmen, Gasser Olivier, Brander Christian, Hess Christoph, Günthard Huldrych F., Brumme Zabrina L., Brumme Chanson J., Bazner Suzane, Rychert Jenna, Tinsley Jake P., Mayer Ken H., Rosenberg Eric, Pereyra Florencia, Levin Joshua Z., Young Sarah K., Jessen Heiko, Altfeld Marcus, Birren Bruce W., Walker Bruce D., and Allen Todd M.. Whole Genome Deep Sequencing of HIV-1 Reveals the Impact of Early Minor Variants Upon Immune Recognition During Acute Infection. PLOS Pathogens, 8(3):e1002529, March 2012. - PMC - PubMed
-
- Holmes Edward. Novel 2019 coronavirus genome, https://virological.org/t/novel-2019-coronavirus-genome/319/1. January 2020.
-
- Wu Fan, Zhao Su, Yu Bin, Chen Yan-Mei, Wang Wen, Song Zhi-Gang, Hu Yi, Tao Zhao-Wu, Tian Jun-Hua, Pei Yuan-Yuan, Yuan Ming-Li, Zhang Yu-Ling, Dai Fa-Hui, Liu Yi, Wang Qi-Min, Zheng Jiao-Jiao, Xu Lin, Holmes Edward C., and Zhang Yong-Zhen. A new coronavirus associated with human respiratory disease in China. Nature, 579(7798):265–269, March 2020. - PMC - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous