

This is a preprint.
Rare germline structural variants increase risk for pediatric solid tumors
- PMID: 38746320
- PMCID: PMC11092455
- DOI: 10.1101/2024.04.27.591484
Rare germline structural variants increase risk for pediatric solid tumors
Update in
-
Rare germline structural variants increase risk for pediatric solid tumors.Science. 2025 Jan 3;387(6729):eadq0071. doi: 10.1126/science.adq0071. Epub 2025 Jan 3. Science. 2025. PMID: 39745975 Free PMC article.
Abstract
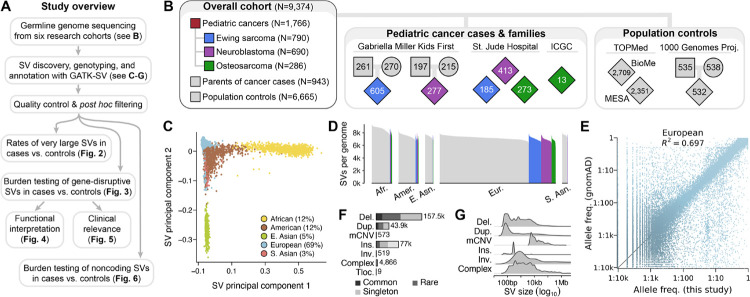
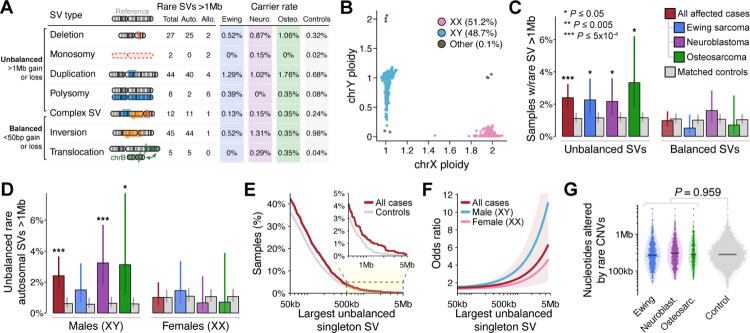
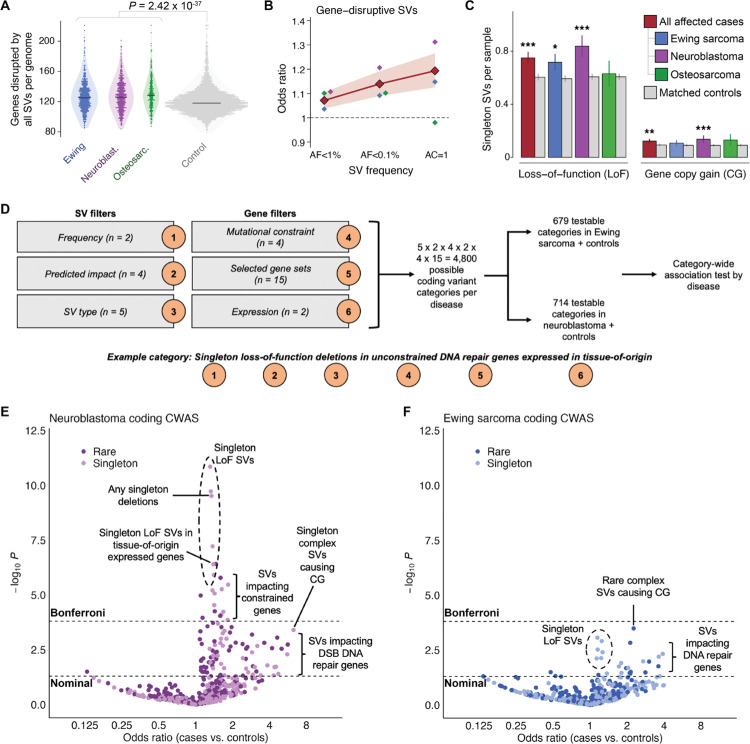
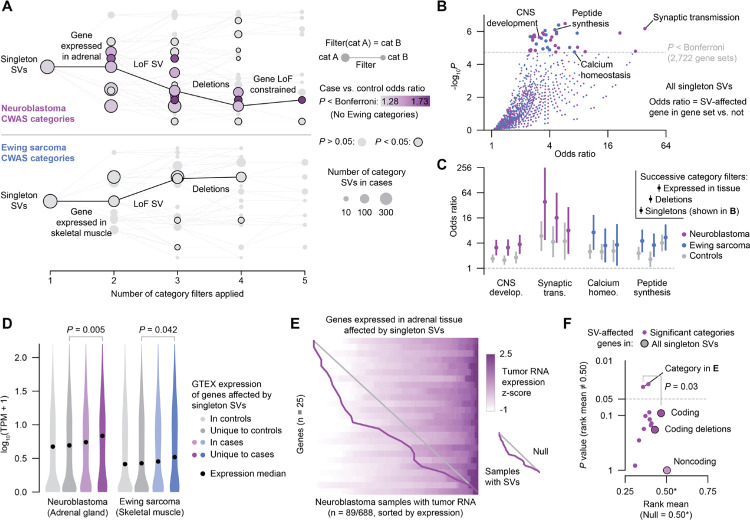
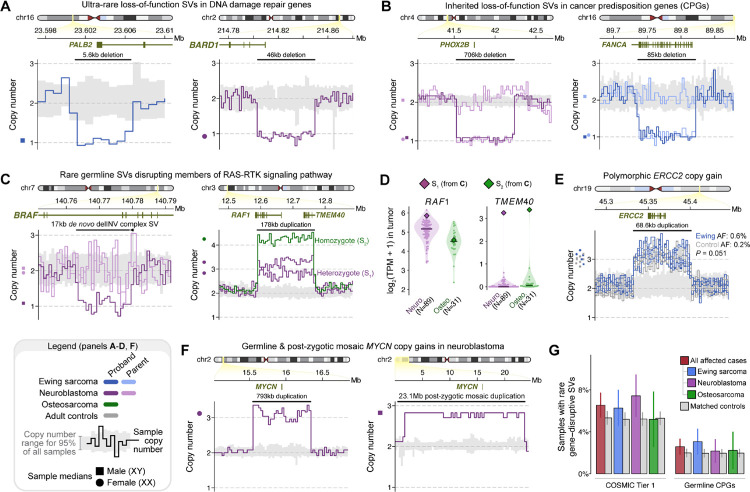
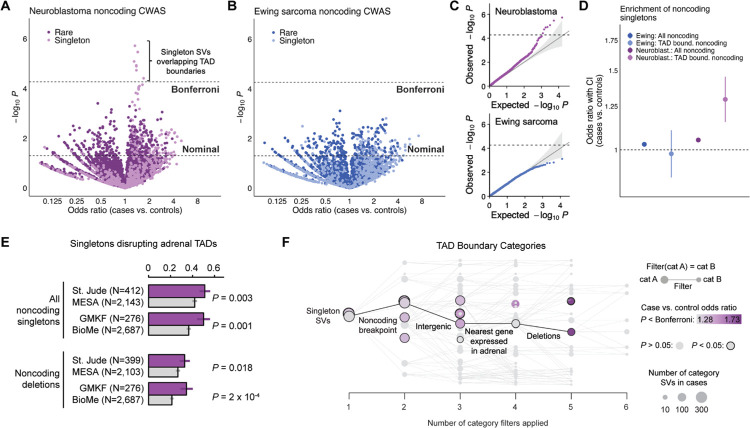
Pediatric solid tumors are rare malignancies that represent a leading cause of death by disease among children in developed countries. The early age-of-onset of these tumors suggests that germline genetic factors are involved, yet conventional germline testing for short coding variants in established predisposition genes only identifies pathogenic events in 10-15% of patients. Here, we examined the role of germline structural variants (SVs)-an underexplored form of germline variation-in pediatric extracranial solid tumors using germline genome sequencing of 1,766 affected children, their 943 unaffected relatives, and 6,665 adult controls. We discovered a sex-biased association between very large (>1 megabase) germline chromosomal abnormalities and a four-fold increased risk of solid tumors in male children. The overall impact of germline SVs was greatest in neuroblastoma, where we revealed burdens of ultra-rare SVs that cause loss-of-function of highly expressed, mutationally intolerant, neurodevelopmental genes, as well as noncoding SVs predicted to disrupt three-dimensional chromatin domains in neural crest-derived tissues. Collectively, our results implicate rare germline SVs as a predisposing factor to pediatric solid tumors that may guide future studies and clinical practice.
Figures
References
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources