

Long-term Remissions Following CD20-Directed Chimeric Antigen Receptor-Adoptive T-cell Therapy
- PMID: 38747505
- PMCID: PMC11215399
- DOI: 10.1158/2643-3230.BCD-23-0263
Long-term Remissions Following CD20-Directed Chimeric Antigen Receptor-Adoptive T-cell Therapy
Abstract
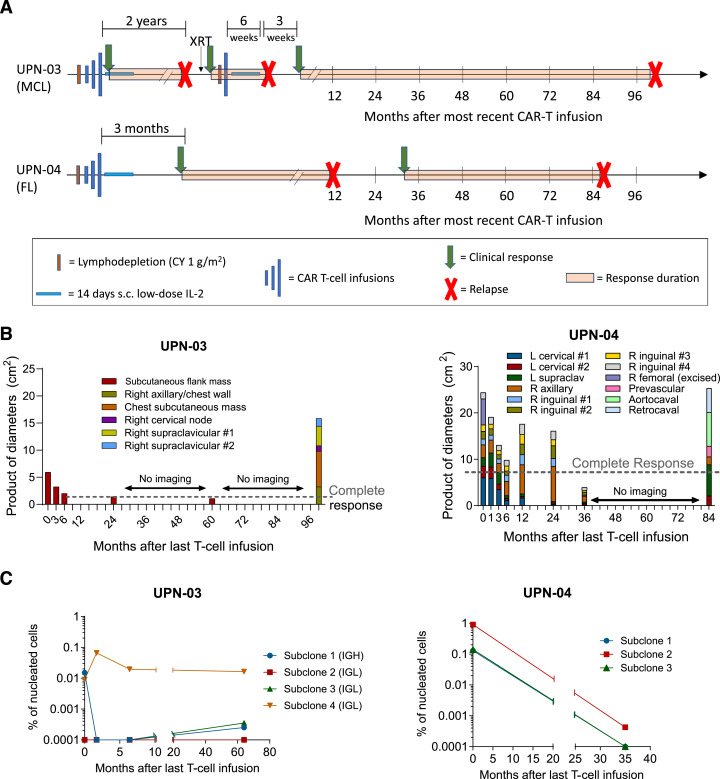
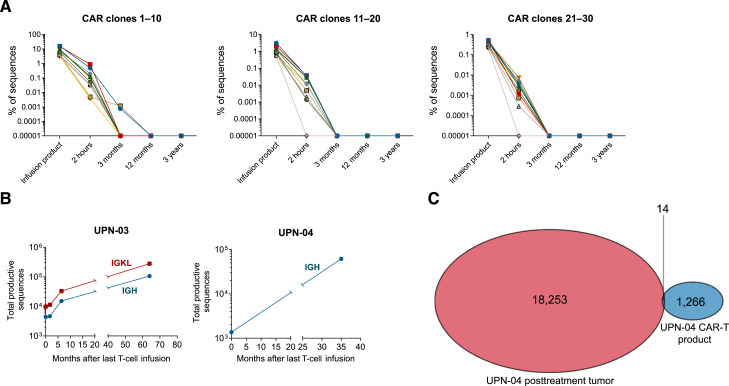
Chimeric antigen receptor (CAR) T-cell therapy produces high response rates in refractory B-cell non-Hodgkin lymphoma, but long-term data are minimal to date. In this study, we present long-term follow-up of a pilot trial testing a CD20-targeting third-generation CAR in patients with relapsed B-cell lymphomas following cyclophosphamide-only lymphodepletion. Two of the three patients in the trial, with mantle cell lymphoma and follicular lymphoma, had remissions lasting more than 7 years, though they ultimately relapsed. The absence of B-cell aplasia in both patients suggested a lack of functional CAR T-cell persistence, leading to the hypothesis that endogenous immune responses were responsible for these long-term remissions. Correlative immunologic analyses supported this hypothesis, with evidence of new humoral and cellular antitumor immune responses proximal to clinical response time points. Collectively, our results suggest that CAR T-cell therapy may facilitate epitope spreading and endogenous immune response formation in lymphomas. Significance: Two of three patients treated with CD20-targeted CAR T-cell therapy had long-term remissions, with evidence of endogenous antitumor immune response formation. Further investigation is warranted to develop conditions that promote epitope spreading in lymphomas.
©2024 American Association for Cancer Research.
Conflict of interest statement
I.R. Kirsch reports personal fees and other support from Adaptive Biotechnologies during the conduct of the study, as well as personal fees and other support from Adaptive Biotechnologies outside the submitted work. R. Gottardo reports other support from Ozette Technologies, Takeda, Sanofi, and Arcellx and grants from 10X Genomics and Owkin outside the submitted work, as well as a patent for markers, methods and systems for identifying cell populations, and diagnosing, monitoring, predicting, and treating conditions issued. K.S. Smythe reports other support from Exicure and X4 Pharmaceuticals and personal fees and other support from Sensei Bio outside the submitted work. A. Greenbaum reports grants from ASH during the conduct of the study. D.J. Green reports grants and other support from Juno Therapeutics, personal fees from GlaxoSmithKline and Ensoma Therapeutics, grants and personal fees from Janssen Biotech, and grants from SpringWorks Therapeutics, Sanofi, Seattle Genetics, Cellectar Biosciences, and Celgene outside the submitted work, as well as a patent for 62/582,270 issued to the Fred Hutchinson Cancer Center and a patent for 62/582,308 issued to Juno Therapeutics. D.G. Maloney reports personal fees from Genentech during the conduct of the study; grants and personal fees from Bristol Myers Squibb, Celgene, Juno Therapeutics, and Kite Pharma, grants from Legend Biotech, personal fees from Caribou Biosciences, Janssen, Chimeric Therapeutics, Bristol Myers Squibb, Genentech, Gilead, and Novartis, and personal fees and other support from A2 Biotherapeutics and NAVAN Technologies outside the submitted work; and a patent for Juno Therapeutics and Bristol Myers Squibb licensed and with royalties paid. B.G. Till reports grants from the Damon Runyon Cancer Research Foundation, Fred Hutchinson Cancer Center, David and Patricia Giuliani Family Foundation, and NIH during the conduct of the study; grants and personal fees from Mustang Bio, grants from Bristol Myers Squibb and Juno, and personal fees from Proteios Technology outside the submitted work; and a patent for Mustang Bio issued, licensed, and with royalties paid. No disclosures were reported by the other authors.
Figures

References
-
- Jacobson CA, Chavez JC, Sehgal AR, William BM, Munoz J, Salles G, et al. . Axicabtagene ciloleucel in relapsed or refractory indolent non-Hodgkin lymphoma (ZUMA-5): a single-arm, multicentre, phase 2 trial. Lancet Oncol 2022;23:91–103. - PubMed
-
- Fowler NH, Dickinson M, Dreyling M, Martinez-Lopez J, Kolstad A, Butler J, et al. . Tisagenlecleucel in adult relapsed or refractory follicular lymphoma: the phase 2 ELARA trial. Nat Med 2022;28:325–32. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
