

A Functional Survey of the Regulatory Landscape of Estrogen Receptor-Positive Breast Cancer Evolution
- PMID: 38753319
- PMCID: PMC11372371
- DOI: 10.1158/2159-8290.CD-23-1157
A Functional Survey of the Regulatory Landscape of Estrogen Receptor-Positive Breast Cancer Evolution
Abstract
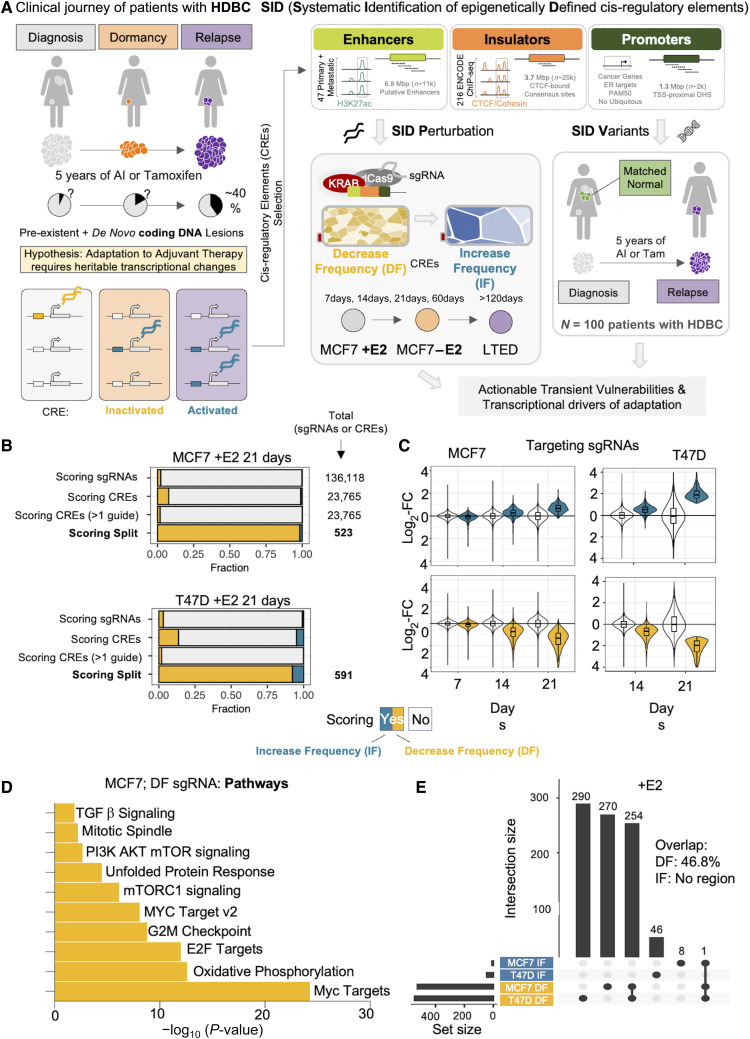
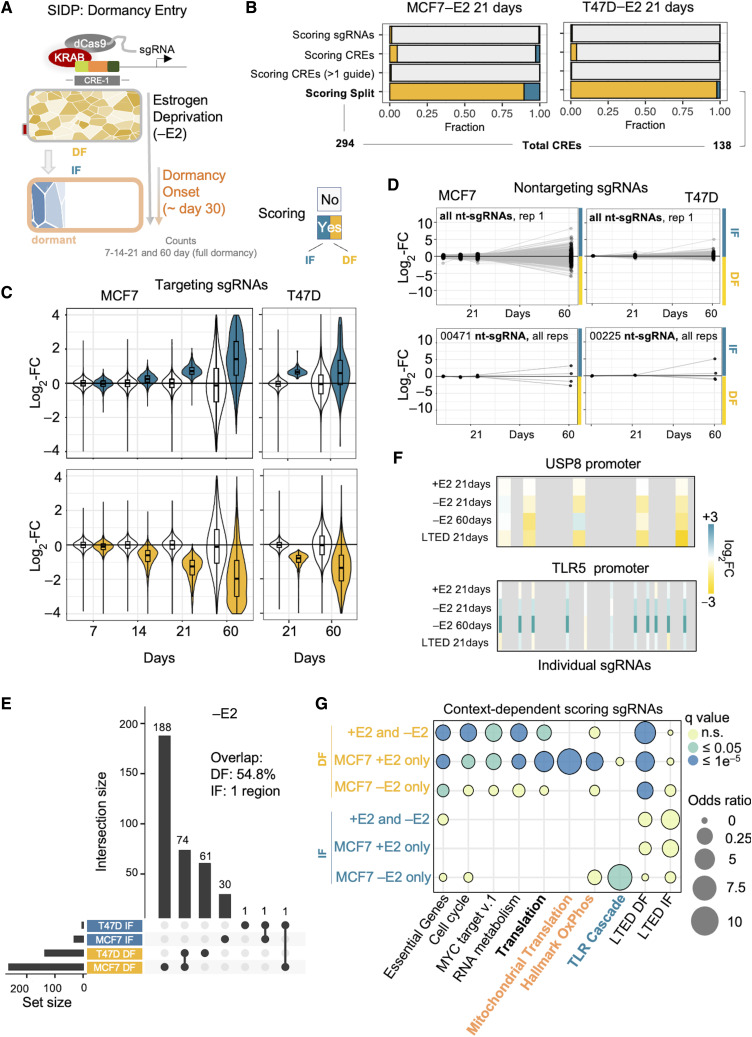
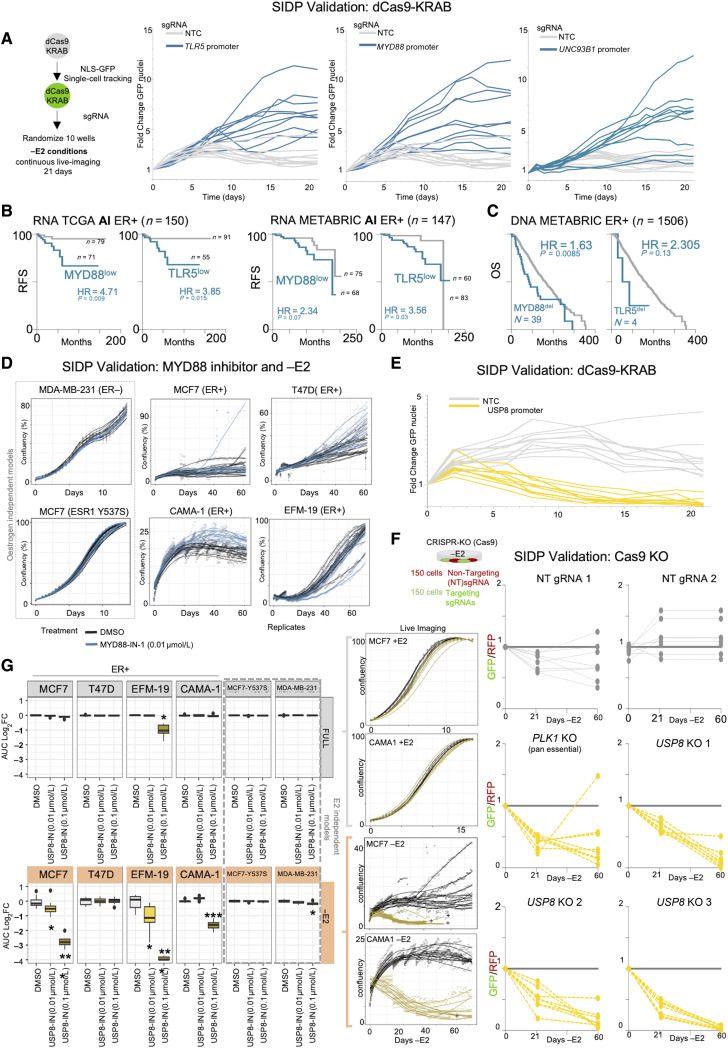
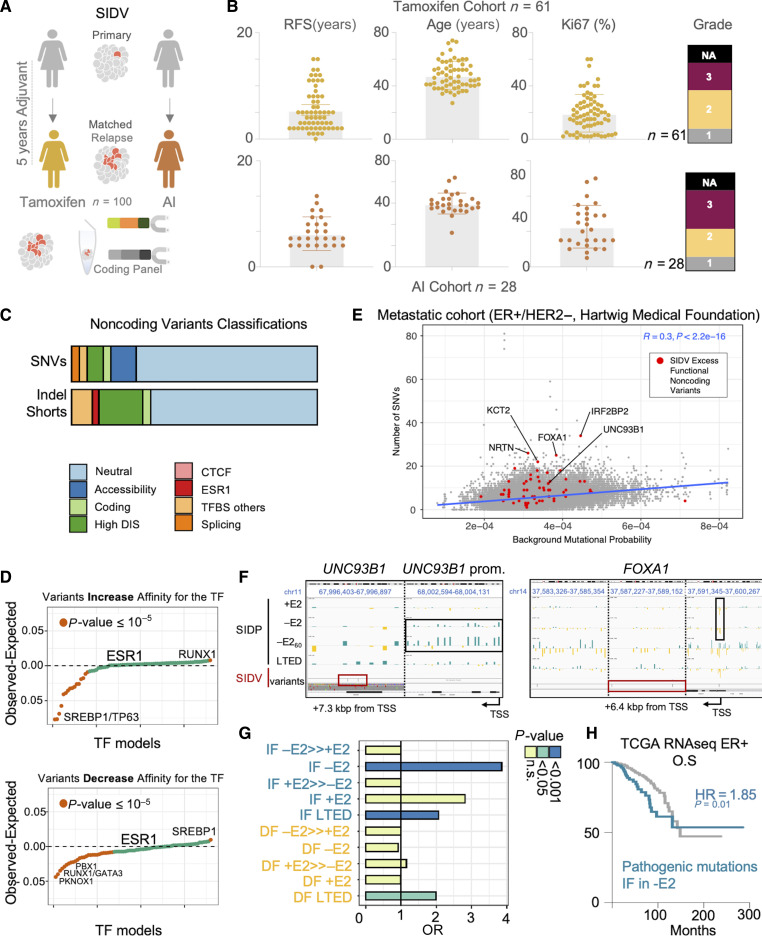
Only a handful of somatic alterations have been linked to endocrine therapy resistance in hormone-dependent breast cancer, potentially explaining ∼40% of relapses. If other mechanisms underlie the evolution of hormone-dependent breast cancer under adjuvant therapy is currently unknown. In this work, we employ functional genomics to dissect the contribution of cis-regulatory elements (CRE) to cancer evolution by focusing on 12 megabases of noncoding DNA, including clonal enhancers, gene promoters, and boundaries of topologically associating domains. Parallel epigenetic perturbation (CRISPRi) in vitro reveals context-dependent roles for many of these CREs, with a specific impact on dormancy entrance and endocrine therapy resistance. Profiling of CRE somatic alterations in a unique, longitudinal cohort of patients treated with endocrine therapies identifies a limited set of noncoding changes potentially involved in therapy resistance. Overall, our data uncover how endocrine therapies trigger the emergence of transient features which could ultimately be exploited to hinder the adaptive process. Significance: This study shows that cells adapting to endocrine therapies undergo changes in the usage or regulatory regions. Dormant cells are less vulnerable to regulatory perturbation but gain transient dependencies which can be exploited to decrease the formation of dormant persisters.
©2024 The Authors; Published by the American Association for Cancer Research.
Conflict of interest statement
M.V. Dieci reports personal fees from Eli Lilly, Novartis, Pfizer, Roche, Gilead, Seagen, Daiichi Sankyo, AstraZeneca, MSD, and Exact Sciences outside the submitted work, as well as a patent for EP20382679.7 licensed to Università di Padova. G. Pruneri reports grants from Fondazione AIRC per la Ricerca sul Cancro ETS - Project ID: 26320 PI: G. Pruneri during the conduct of the study, as well as personal fees from Novartis, Illumina, and Eli Lilly and Company outside the submitted work. G.G. Galli reports being an employee and a shareholder of Novartis. No disclosures were reported by the other authors.
Figures
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
